Entry Database : PDB / ID : 2x2qTitle Crystal structure of an 'all locked' LNA duplex at 1.9 angstrom resolution (LOCKED NUCLEIC ACID DERIVED FROM TRNA SER ACCEPTOR STEM ...) x 2 Keywords / / / Function / homology / / Biological species SYNTHETIC CONSTRUCT (others) Method / / / Resolution : 1.9 Å Authors Eichert, A. / Behling, K. / Fuerste, J.P. / Betzel, C. / Erdmann, V.A. / Foerster, C. Journal : Nucleic Acids Res. / Year : 2010Title : The Crystal Structure of an 'All Locked' Nucleic Acid Duplex.Authors : Eichert, A. / Behling, K. / Betzel, C. / Erdmann, V.A. / Furste, J.P. / Forster, C. History Deposition Jan 15, 2010 Deposition site / Processing site Revision 1.0 Feb 16, 2011 Provider / Type Revision 1.1 May 7, 2011 Group Revision 1.2 Jul 13, 2011 Group Revision 1.3 Jul 12, 2017 Group Category pdbx_struct_conn_angle / struct_conn ... pdbx_struct_conn_angle / struct_conn / struct_site / struct_site_gen Item _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id ... _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_symmetry / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.conn_type_id / _struct_conn.id / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn.ptnr2_symmetry Revision 1.4 Dec 20, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr1_symmetry / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.ptnr3_symmetry / _pdbx_struct_conn_angle.value / _struct_conn.conn_type_id / _struct_conn.id / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn.ptnr2_symmetry
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords DNA / MODIFIED NUCLEIC ACID /
DNA / MODIFIED NUCLEIC ACID /  Function and homology information
Function and homology information Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj






 Assembly
Assembly


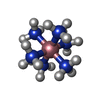

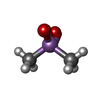

 Mass: 161.116 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CoH18N6
Mass: 161.116 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CoH18N6 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2
Mass: 136.989 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6AsO2 Sample preparation
Sample preparation / Beamline: 5.2R / Wavelength: 1
/ Beamline: 5.2R / Wavelength: 1  Processing
Processing