Journal: Cell / Year: 2008 Title: Fiber formation across the bacterial outer membrane by the chaperone/usher pathway. Authors: Han Remaut / Chunyan Tang / Nadine S Henderson / Jerome S Pinkner / Tao Wang / Scott J Hultgren / David G Thanassi / Gabriel Waksman / Huilin Li / Abstract: Gram-negative pathogens commonly exhibit adhesive pili on their surfaces that mediate specific attachment to the host. A major class of pili is assembled via the chaperone/usher pathway. Here, the ...Gram-negative pathogens commonly exhibit adhesive pili on their surfaces that mediate specific attachment to the host. A major class of pili is assembled via the chaperone/usher pathway. Here, the structural basis for pilus fiber assembly and secretion performed by the outer membrane assembly platform--the usher--is revealed by the crystal structure of the translocation domain of the P pilus usher PapC and single particle cryo-electron microscopy imaging of the FimD usher bound to a translocating type 1 pilus assembly intermediate. These structures provide molecular snapshots of a twinned-pore translocation machinery in action. Unexpectedly, only one pore is used for secretion, while both usher protomers are used for chaperone-subunit complex recruitment. The translocating pore itself comprises 24 beta strands and is occluded by a folded plug domain, likely gated by a conformationally constrained beta-hairpin. These structures capture the secretion of a virulence factor across the outer membrane of gram-negative bacteria.
SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 47-STRANDED BARREL THIS IS REPRESENTED BY A 48-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords TRANSMEMBRANE / OUTER MEMBRANE / USHER / P PILUS /
TRANSMEMBRANE / OUTER MEMBRANE / USHER / P PILUS /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj Assembly
Assembly


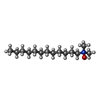
 Mass: 229.402 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM
Mass: 229.402 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM
 Mass: 306.438 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H34O5 / Comment: C8E, detergent*YM
Mass: 306.438 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H34O5 / Comment: C8E, detergent*YM Mass: 18.015 Da / Num. of mol.: 34 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 34 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.979
/ Beamline: ID29 / Wavelength: 0.979  Processing
Processing