 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2vn1: Crystal structure of the FK506-binding domain of Plasmodium falci... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2vn1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the FK506-binding domain of Plasmodium falciparum FKBP35 in complex with FK506 | ||||||
 Components Components | 70 KDA PEPTIDYLPROLYL ISOMERASE | ||||||
 Keywords Keywords |  ISOMERASE / FKBP / FK506 / TPR REPEAT / PLASMODIUM FALCIPARUM ISOMERASE / FKBP / FK506 / TPR REPEAT / PLASMODIUM FALCIPARUM | ||||||
| Function / homology |  Function and homology information Function and homology informationHSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / negative regulation of phosphoprotein phosphatase activity / FK506 binding / protein peptidyl-prolyl isomerization / chaperone-mediated protein folding / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / protein folding / protein dimerization activity / nucleus / cytoplasmSimilarity search - Function | ||||||
| Biological species |  PLASMODIUM FALCIPARUM (malaria parasite P. falciparum) PLASMODIUM FALCIPARUM (malaria parasite P. falciparum) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.35 Å | ||||||
 Authors Authors | Kotaka, M. / Alag, R. / Ye, H. / Preiser, P.R. / Yoon, H.S. / Lescar, J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2008 Title: Crystal Structure of the Fk506 Binding Domain of Plasmodium Falciparum Fkbp35 in Complex with Fk506. Authors: Kotaka, M. / Ye, H. / Alag, R. / Hu, G. / Bozdech, Z. / Preiser, P.R. / Yoon, H.S. / Lescar, J. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2vn1.cif.gz | 68.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2vn1.ent.gz | 50.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2vn1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vn/2vn1ftp://data.pdbj.org/pub/pdb/validation_reports/vn/2vn1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1yatS S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.7224, -0.6915, 0.0035), Vector : |
-Components
| #1: Protein | Mass: 14531.420 Da / Num. of mol.: 2 / Fragment: FK506-BINDING DOMAIN, RESIDUES 1-127 Source method: isolated from a genetically manipulated source Source: (gene. exp.) PLASMODIUM FALCIPARUM (malaria parasite P. falciparum)Strain: 3D7 / Plasmid: PET29B / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q8I4V8, peptidylprolyl isomerase ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q8I4V8, peptidylprolyl isomerase#2: Chemical | Tacrolimus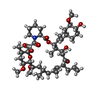  Mass: 804.018 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C44H69NO12 / Comment: medication*YM Mass: 804.018 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C44H69NO12 / Comment: medication*YM#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 162 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 162 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.97 Å3/Da / Density % sol: 58.6 % / Description: NONE |
|---|---|
| Crystal grow | pH: 5 / Details: 2.7M NA MALONATE, PH5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.933 / Beamline: ID14-1 / Wavelength: 0.933 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jun 24, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 2.35→30 Å / Num. obs: 14883 / % possible obs: 99.8 % / Observed criterion σ(I): 0 / Redundancy: 20.7 % / Biso Wilson estimate: 30.5 Å2 / Rmerge(I) obs: 0.1 / Net I/σ(I): 41.1 |
| Reflection shell | Resolution: 2.35→2.43 Å / Rmerge(I) obs: 0.38 / Mean I/σ(I) obs: 8.5 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1YAT Resolution: 2.35→29.65 Å / Rfactor Rfree error: 0.009 / Data cutoff high absF: 1800752.84 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 40.8564 Å2 / ksol: 0.4 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.35→29.65 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.35→2.5 Å / Rfactor Rfree error: 0.033 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
