 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2veo: X-ray structure of Candida antarctica lipase A in its closed state. -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2veo | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray structure of Candida antarctica lipase A in its closed state. | ||||||
 Components Components | LIPASE A | ||||||
 Keywords Keywords |  HYDROLASE / LIPASE / INTERFACIAL ACTIVATION / SUBSTRATE SPECIFICITY HYDROLASE / LIPASE / INTERFACIAL ACTIVATION / SUBSTRATE SPECIFICITY | ||||||
| Function / homology |  Function and homology informationtriacylglycerol lipase / triglyceride lipase activity / lipid catabolic process / extracellular region Function and homology informationtriacylglycerol lipase / triglyceride lipase activity / lipid catabolic process / extracellular regionSimilarity search - Function | ||||||
| Biological species |  PSEUDOZYMA ANTARCTICA (fungus) PSEUDOZYMA ANTARCTICA (fungus) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 2.2 Å | ||||||
 Authors Authors | Ericsson, D.J. / Kasrayan, A. / Johansson, P. / Bergfors, T. / Sandstrom, A.G. / Backvall, J.E. / Mowbray, S.L. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2008 Title: X-Ray Structure of Candida Antarctica Lipase a Shows a Novel Lid Structure and a Likely Mode of Interfacial Activation. Authors: Ericsson, D.J. / Kasrayan, A. / Johansson, P. / Bergfors, T. / Sandstrom, A.G. / Backvall, J.E. / Mowbray, S.L. | ||||||
| History |
| ||||||
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR PROVIDED. | ||||||
| Remark 700 | SHEET DETERMINATION METHOD: AUTHOR PROVIDED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2veo.cif.gz | 171.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2veo.ent.gz | 143 KB | Display | PDB format |
| PDBx/mmJSON format | 2veo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ve/2veoftp://data.pdbj.org/pub/pdb/validation_reports/ve/2veo | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|

-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.7025, -0.7068, 0.0837), Vector : |
-Components
| #1: Protein | Mass: 47201.984 Da / Num. of mol.: 2 / Fragment: RESIDUES 88-528 Source method: isolated from a genetically manipulated source Source: (gene. exp.) PSEUDOZYMA ANTARCTICA (fungus)Description: CANDIDA ANTARCTICA RECLASSIFIED AS PSEUDOZYMA APHIDIS IN 2006. Plasmid: PPICZALPHA-C / Production host: PICHIA PASTORIS (fungus) / Strain (production host): X33 / References: UniProt: W3VKA4, triacylglycerol lipase#2: Chemical | ChemComp-IUM / Uranyl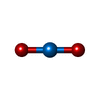  Mass: 270.028 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: O2U Mass: 270.028 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: O2U#3: Chemical | ChemComp-PG4 / | Polyethylene glycol  Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#4: Chemical | ChemComp-GOL / | Glycerol  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 466 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 466 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | URANYL (VI) ION (IUM): URANYL OXYGENS IMPOSSIBLE | Sequence details | PRIMARY AMINO ACID SEQUENCE UNAVAILABL | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.2 Å3/Da / Density % sol: 60 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.4 Details: 2 MICROLITER 10 MILLIGRAM/MILLILITER PROTEIN IN 0.002M TRIS-HCL, PH 8.0 MIXED WITH 1 MICROLITER 0.2M AMMONIUM SULFATE, 0.1M BIS-TRIS, PH 5.5, AND 25% (W/V) PEG 3350. |
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.9537 / Beamline: ID29 / Wavelength: 0.9537 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: May 15, 2006 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9537 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→30 Å / Num. obs: 63098 / % possible obs: 96 % / Observed criterion σ(I): 2 / Redundancy: 4 % / Biso Wilson estimate: 21.7 Å2 / Rmerge(I) obs: 0.14 / Net I/σ(I): 10.2 |
| Reflection shell | Resolution: 2.2→2.32 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.62 / Mean I/σ(I) obs: 2 / % possible all: 85.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIRAS Starting model: NONE Resolution: 2.2→30 Å / Cor.coef. Fo:Fc: 0.932 / Cor.coef. Fo:Fc free: 0.905 / SU B: 3.698 / SU ML: 0.095 / Cross valid method: THROUGHOUT / ESU R: 0.2 / ESU R Free: 0.169 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.73 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|