 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
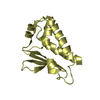
Yorodumi- PDB-2qh7: MitoNEET is a uniquely folded 2Fe-2S outer mitochondrial membrane... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2qh7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | MitoNEET is a uniquely folded 2Fe-2S outer mitochondrial membrane protein stabilized by pioglitazone | ||||||
 Components Components | Zinc finger CDGSH-type domain 1 | ||||||
 Keywords Keywords | METAL BINDING PROTEIN /  MitoNEET / 2Fe-2S protein / Outer mitochrodrial membrane protein / pioglitazone binding MitoNEET / 2Fe-2S protein / Outer mitochrodrial membrane protein / pioglitazone binding | ||||||
| Function / homology |  Function and homology information Function and homology informationprotein maturation by [2Fe-2S] cluster transfer / cysteine transaminase / L-cysteine transaminase activity / cytoplasmic side of mitochondrial outer membrane / regulation of cellular respiration / regulation of autophagy / 2 iron, 2 sulfur cluster binding / pyridoxal phosphate binding / mitochondrial outer membrane / membrane => GO:0016020 ...protein maturation by [2Fe-2S] cluster transfer / cysteine transaminase / L-cysteine transaminase activity / cytoplasmic side of mitochondrial outer membrane / regulation of cellular respiration / regulation of autophagy / 2 iron, 2 sulfur cluster binding / pyridoxal phosphate binding / mitochondrial outer membrane / membrane => GO:0016020 / protein homodimerization activity / mitochondrion / identical protein binding / metal ion bindingSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.5 Å | ||||||
 Authors Authors | Paddock, M.L. / Wiley, S.E. / Axelrod, H.L. / Cohen, A.E. / Roy, M. / Abresch, E.C. / Capraro, D. / Murphy, A.N. / Nechushtai, R. / Dixon, J.E. / Jennings, P.A. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.Usa / Year: 2007 Title: MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Authors: Paddock, M.L. / Wiley, S.E. / Axelrod, H.L. / Cohen, A.E. / Roy, M. / Abresch, E.C. / Capraro, D. / Murphy, A.N. / Nechushtai, R. / Dixon, J.E. / Jennings, P.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2qh7.cif.gz | 45.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2qh7.ent.gz | 30.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2qh7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qh/2qh7ftp://data.pdbj.org/pub/pdb/validation_reports/qh/2qh7 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Refine code: 6
|
-Components
| #1: Protein | Mass: 8948.327 Da / Num. of mol.: 2 / Fragment: water-solule domain of MitoNEET-residues 33-108 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ZCD1 / Production host:  Escherichia coli (E. coli) / References: UniProt: Q1X902, UniProt: Q9NZ45*PLUS Escherichia coli (E. coli) / References: UniProt: Q1X902, UniProt: Q9NZ45*PLUS#2: Chemical | Iron–sulfur cluster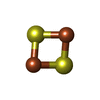  Mass: 175.820 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe2S2 Mass: 175.820 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe2S2#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 128 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 128 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.91 Å3/Da / Density % sol: 35.73 % |
|---|
-Data collection
| Diffraction |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | D res high: 1.8 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.5→38 Å / Num. obs: 21479 / % possible obs: 94.7 % / Biso Wilson estimate: 27.683 Å2 / Rmerge(I) obs: 0.058 / Net I/σ(I): 30.92 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1,2
|

-Phasing
| Phasing | Method: MAD | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing set |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MAD set |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing MAD set site |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm | FOM : 0.73 / FOM acentric: 0.74 / FOM centric: 0.71 / Reflection: 13466 / Reflection acentric: 11564 / Reflection centric: 1902 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.5→38 Å / Cor.coef. Fo:Fc: 0.972 / Cor.coef. Fo:Fc free: 0.954 / SU B: 4.564 / SU ML: 0.078 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.078 / ESU R Free: 0.084 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. 3.ELECTRON DENSITIES CORRESPONDING TO RESIDUES 33-41 AND 107-108 ON THE A SUBUNIT AND ...Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. 3.ELECTRON DENSITIES CORRESPONDING TO RESIDUES 33-41 AND 107-108 ON THE A SUBUNIT AND RESIDUES 33-42 AND 108 ON THE B SUBUNIT WERE DISORDERED AND THESE RESIDUES WERE NOT MODELED. 4.A 2FE-2S CLUSTER (FES) WAS MODELED INTO EACH SUBUNIT IN THE ASYMMETRIC UNIT. THE PRESENCE OF THE 2FE-2S CLUSTER WAS CORRBORATED BY ANOMALOUS DIFFERENCE MAPS. THE PROTEIN LIGANDS TO THE FE ATOMS IN THE 2FE-2S CLUSTERS ARE CYS 72, CYS 74, CYS 83, AND HIS 87.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 33.279 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→38 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Number: 917 / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.542 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
