 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2q9p: Human diphosphoinositol polyphosphate phosphohydrolase 1, Mg-F complex -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2q9p | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human diphosphoinositol polyphosphate phosphohydrolase 1, Mg-F complex | ||||||
 Components Components | Diphosphoinositol polyphosphate phosphohydrolase 1 | ||||||
 Keywords Keywords |  HYDROLASE / DIPHOSPHOINOSITOL POLYPHOSPHATE PHOSPHOHYDROLASE / NUDIX / INOSITOL PYROPHOSPHATE METABOLISM / STRUCTURAL GENOMICS CONSORTIUM / SGC HYDROLASE / DIPHOSPHOINOSITOL POLYPHOSPHATE PHOSPHOHYDROLASE / NUDIX / INOSITOL PYROPHOSPHATE METABOLISM / STRUCTURAL GENOMICS CONSORTIUM / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationdiphosphoinositol polyphosphate catabolic process / inositol diphosphate pentakisphosphate diphosphatase activity / inositol-3,5-bisdiphosphate-2,3,4,6-tetrakisphosphate 5-diphosphatase activity / inositol diphosphate tetrakisphosphate diphosphatase activity / inositol-5-diphosphate-1,2,3,4,6-pentakisphosphate diphosphatase activity / endopolyphosphatase / diadenosine polyphosphate catabolic process / 5'-(N7-methyl 5'-triphosphoguanosine)-[mRNA] diphosphatase / endopolyphosphatase activity / bis(5'-adenosyl)-hexaphosphatase activity ...diphosphoinositol polyphosphate catabolic process / inositol diphosphate pentakisphosphate diphosphatase activity / inositol-3,5-bisdiphosphate-2,3,4,6-tetrakisphosphate 5-diphosphatase activity / inositol diphosphate tetrakisphosphate diphosphatase activity / inositol-5-diphosphate-1,2,3,4,6-pentakisphosphate diphosphatase activity / endopolyphosphatase / diadenosine polyphosphate catabolic process / 5'-(N7-methyl 5'-triphosphoguanosine)-[mRNA] diphosphatase / endopolyphosphatase activity / bis(5'-adenosyl)-hexaphosphatase activity / diphosphoinositol polyphosphate metabolic process / RNA decapping / diadenosine pentaphosphate catabolic process / diadenosine hexaphosphate catabolic process / adenosine 5'-(hexahydrogen pentaphosphate) catabolic process / diphosphoinositol-polyphosphate diphosphatase / diphosphoinositol-polyphosphate diphosphatase activity / 5'-(N(7)-methyl 5'-triphosphoguanosine)-[mRNA] diphosphatase activity / diadenosine hexaphosphate hydrolase (ATP-forming) / Synthesis of pyrophosphates in the cytosol / bis(5'-adenosyl)-pentaphosphatase activity / 5'-(N7-methylguanosine 5'-triphospho)-[mRNA] hydrolase / 5'-(N(7)-methylguanosine 5'-triphospho)-[mRNA] hydrolase activity / cell-cell signaling / manganese ion binding / magnesium ion binding / zinc ion binding / nucleus / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.65 Å | ||||||
 Authors Authors | Thorsell, A.G. / Busam, R. / Arrowsmith, C.H. / Berglund, H. / Collins, R. / Dahlgren, L.G. / Edwards, A. / Flodin, S. / Flores, A. / Graslund, S. ...Thorsell, A.G. / Busam, R. / Arrowsmith, C.H. / Berglund, H. / Collins, R. / Dahlgren, L.G. / Edwards, A. / Flodin, S. / Flores, A. / Graslund, S. / Hammarstrom, M. / Holmberg-Schiavone, L. / Johansson, I. / Kallas, A. / Karlberg, T. / Kotenyova, T. / Lehtio, L. / Moche, M. / Nordlund, P. / Nyman, T. / Ogg, D. / Sagemark, J. / Sundstrom, M. / Van den Berg, S. / Weigelt, J. / Welin, M. / Persson, C. / Hallberg, B.M. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Proteins / Year: 2009 Title: Crystal structure of human diphosphoinositol phosphatase 1. Authors: Thorsell, A.G. / Persson, C. / Graslund, S. / Hammarstrom, M. / Busam, R.D. / Hallberg, B.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2q9p.cif.gz | 51.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2q9p.ent.gz | 33.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2q9p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q9/2q9pftp://data.pdbj.org/pub/pdb/validation_reports/q9/2q9p | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2fvvSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 22055.725 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Tissue: Uterus / Gene: NUDT3, DIPP, DIPP1 / Plasmid: pNIC-BSA4 / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3)References: UniProt: O95989, diphosphoinositol-polyphosphate diphosphatase |
|---|
-Non-polymers , 5 types, 162 molecules 


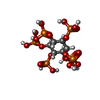

| #2: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-CL / | Chloride Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-F / Fluoride Mass: 18.998 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: F Mass: 18.998 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: F#5: Chemical | ChemComp-IHP / | Phytic acid Mass: 660.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H18O24P6 Mass: 660.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H18O24P6#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 146 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.9 Å3/Da / Density % sol: 35.4 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 4.5 Details: 30% PEG 8000, 200 mM LiCl, 5 mM MgCl2, 20 mM NaF, 5 mM IP6, pH 4.5, VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR / Wavelength: 1.5418 |
| Detector | Type: Bruker Platinum 135 / Detector: CCD / Date: May 19, 2007 / Details: Helios multilayer mirrors |
| Radiation | Monochromator: Helios multilayer / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.65→50 Å / Num. all: 20890 / Num. obs: 20848 / % possible obs: 99.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 2.1 % / Rsym value: 0.054 / Net I/σ(I): 11.7 |
| Reflection shell | Resolution: 1.65→1.75 Å / Mean I/σ(I) obs: 2.1 / Rsym value: 0.461 / % possible all: 99.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB entry 2FVV Resolution: 1.65→32.21 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.944 / SU B: 2.277 / SU ML: 0.077 / Cross valid method: THROUGHOUT / ESU R: 0.099 / ESU R Free: 0.101 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: Structure was determined starting from rigid body refinement of the PDB entry 2FVV with Refmac 5 program. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.789 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.65→32.21 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.65→1.693 Å / Total num. of bins used: 20
|
