 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2q78: Crystal structure of a thioesterase-like protein (tm0581) from th... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2q78 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a thioesterase-like protein (tm0581) from thermotoga maritima msb8 at 2.20 A resolution | ||||||
 Components Components | Uncharacterized protein | ||||||
 Keywords Keywords |  HYDROLASE / Structural genomics / Joint Center for Structural Genomics / JCSG / Protein Structure Initiative / PSI-2 HYDROLASE / Structural genomics / Joint Center for Structural Genomics / JCSG / Protein Structure Initiative / PSI-2 | ||||||
| Function / homology | Fluoroacetyl-CoA thioesterase / Hotdog Thioesterase / Thiol Ester Dehydrase; Chain A / HotDog domain superfamily / Roll / Alpha Beta / MALONYL-COENZYME A / Uncharacterized protein Function and homology information Function and homology information | ||||||
| Biological species |   Thermotoga maritima MSB8 (bacteria) Thermotoga maritima MSB8 (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.2 Å | ||||||
 Authors Authors | Joint Center for Structural Genomics (JCSG) | ||||||
 Citation Citation | Journal: To be published Title: Crystal structure of Uncharacterized protein TM0581 (TM0581) from Thermotoga maritima at 2.20 A resolution Authors: Joint Center for Structural Genomics (JCSG) | ||||||
| History |
| ||||||
| Remark 300 | BIOMOLECULE: 1,2,3,4 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 8 ... BIOMOLECULE: 1,2,3,4 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 8 CHAIN(S). SEE REMARK 350 FOR INFORMATION ON GENERATING THE BIOLOGICAL MOLECULE(S). SIZE EXCLUSION CHROMATOGRAPHY SUPPORTS THE ASSIGNMENT OF A DIMER AS A SIGNIFICANT OLIGOMERIZATION STATE. | ||||||
| Remark 999 | SEQUENCE THE CONSTRUCT WAS EXPRESSED WITH A PURIFICATION TAG MGSDKIHHHHHH FOLLOWED BY THE TARGET SEQUENCE. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2q78.cif.gz | 236.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2q78.ent.gz | 196 KB | Display | PDB format |
| PDBx/mmJSON format | 2q78.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q7/2q78ftp://data.pdbj.org/pub/pdb/validation_reports/q7/2q78 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Beg label comp-ID: ASP / End label comp-ID: GLU / Refine code: 5 / Auth seq-ID: 3 - 130 / Label seq-ID: 15 - 142
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | SIZE EXCLUSION CHROMATOGRAPHY SUPPORTS THE ASSIGNMENT OF A DIMER AS A SIGNIFICANT OLIGOMERIZATION STATE. |
-Components
| #1: Protein | Mass: 17633.559 Da / Num. of mol.: 8 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermotoga maritima MSB8 (bacteria) / Species: Thermotoga maritima / Strain: MSB8, DSM 3109, JCM 10099 / Gene: TM_0581 / Plasmid: speedET / Production host: Escherichia coli (E. coli) / Strain (production host): HK100 / References: UniProt: Q9WZ50#2: Chemical | ChemComp-MLC / Malonyl-CoA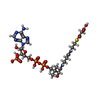  Mass: 853.580 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C24H38N7O19P3S Mass: 853.580 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C24H38N7O19P3S#3: Chemical | Chloride  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 282 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 282 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.96 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: NANODROP, 0.8M Lithium chloride, 10.0% PEG 6000, 0.1M HEPES pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-2 / Wavelength: 0.97913 Å / Beamline: BL9-2 / Wavelength: 0.97913 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: May 13, 2007 / Details: Flat collimating mirror, toroid focusing mirror | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97913 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.2→29.975 Å / Num. obs: 65127 / % possible obs: 99.7 % / Redundancy: 4.8 % / Biso Wilson estimate: 37.8 Å2 / Rmerge(I) obs: 0.112 / Rsym value: 0.112 / Net I/σ(I): 10.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: SAD |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.2→29.975 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.941 / SU B: 10.66 / SU ML: 0.145 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.235 / ESU R Free: 0.193 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. ATOM RECORDS CONTAIN RESIDUAL B FACTORS ONLY. 3. RESIDUES A/B/C/D/E/G131-141 AND F134-141 AND MOST OF THE N_TERMINAL PURIFICATION TAG ...Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. ATOM RECORDS CONTAIN RESIDUAL B FACTORS ONLY. 3. RESIDUES A/B/C/D/E/G131-141 AND F134-141 AND MOST OF THE N_TERMINAL PURIFICATION TAG ARE DISORDERED AND NOT INCLUDED IN THE MODEL. 4. ONE MALONYL-COA (MCL) IS MODELED FOR EACH MONOMER BASED ON DENSITY AND STRUCTURAL HOMOLOGY (PDB ID: 2F3X). THE OCCUPANCY OF OF MCL IN CHAIN H IS LIKELY LESS THAN 1.0. 5. CL MOLECULES FROM THE CRYSTALLIZATION/CRYO SOLUTION ARE MODELED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 43.94 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→29.975 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Refine-ID: X-RAY DIFFRACTION
|