 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ki5: HERPES SIMPLEX TYPE-1 THYMIDINE KINASE IN COMPLEX WITH THE DRUG A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ki5 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | HERPES SIMPLEX TYPE-1 THYMIDINE KINASE IN COMPLEX WITH THE DRUG ACICLOVIR AT 1.9A RESOLUTION | |||||||||
 Components Components | PROTEIN (THYMIDINE KINASE) | |||||||||
 Keywords Keywords |  TRANSFERASE / TRANSFERASE (PHOSPHOTRANSFERASE) THYMID / TRANSFERASE (PHOSPHOTRANSFERASE) THYMIDINE KINASE / VIRIDAE / DS-DNA ENVELOPED VIRUSES / HERPESVIRIDAE / ALPHAHERPESVIRINAE ANTIVIRAL DRUG (ACICLOVIR) TRANSFERASE / TRANSFERASE (PHOSPHOTRANSFERASE) THYMID / TRANSFERASE (PHOSPHOTRANSFERASE) THYMIDINE KINASE / VIRIDAE / DS-DNA ENVELOPED VIRUSES / HERPESVIRIDAE / ALPHAHERPESVIRINAE ANTIVIRAL DRUG (ACICLOVIR) | |||||||||
| Function / homology |  Function and homology information Function and homology informationTMP biosynthetic process / thymidine kinase / thymidine kinase activity / DNA biosynthetic process / phosphorylation / ATP bindingSimilarity search - Function | |||||||||
| Biological species |   Human herpesvirus 1 (Herpes simplex virus type 1) Human herpesvirus 1 (Herpes simplex virus type 1) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | |||||||||
 Authors Authors | Bennett, M.S. / Wien, F. / Champness, J.N. / Batuwangala, T. / Rutherford, T. / Summers, W.C. / Sun, H. / Wright, G. / Sanderson, M.R. | |||||||||
 Citation Citation | Journal: FEBS Lett. / Year: 1999 Title: Structure to 1.9 A resolution of a complex with herpes simplex virus type-1 thymidine kinase of a novel, non-substrate inhibitor: X-ray crystallographic comparison with binding of aciclovir. Authors: Bennett, M.S. / Wien, F. / Champness, J.N. / Batuwangala, T. / Rutherford, T. / Summers, W.C. / Sun, H. / Wright, G. / Sanderson, M.R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ki5.cif.gz | 136.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ki5.ent.gz | 104.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2ki5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ki/2ki5ftp://data.pdbj.org/pub/pdb/validation_reports/ki/2ki5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1qhiC  1kimS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39862.570 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: ACICLOVIR, ANTIVIRAL DRUG AS DEPOSITED IN 1KI5 Source: (gene. exp.) Human herpesvirus 1 (Herpes simplex virus type 1)Genus: Simplexvirus / Strain: SY211 / Gene: TK / Plasmid: PT7:HSVTK / Gene (production host): TK / Production host:  Escherichia coli (E. coli) / Strain (production host): SY211 Escherichia coli (E. coli) / Strain (production host): SY211References: UniProt: P03176, UniProt: P0DTH5*PLUS, thymidine kinase#2: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | Aciclovir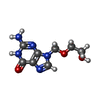  Mass: 225.205 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H11N5O3 / Comment: medication, antivirus*YM Mass: 225.205 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H11N5O3 / Comment: medication, antivirus*YM#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 292 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 292 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.8 % | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: CRYSTALLIZATION CONDITIONS: PUBLISHED IN BENNETT, M.S. ET AL. FEBS LETT., VOL.443 (1999) 121-125, pH 7.5 | |||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 25 ℃ / Method: vapor diffusion | |||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.9 / Beamline: BM14 / Wavelength: 0.9 |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Apr 1, 1998 / Details: MIRRORS |
| Radiation | Monochromator: OTWINOWSKI MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 1.82→16 Å / Num. obs: 63458 / % possible obs: 97 % / Observed criterion σ(I): 3 / Redundancy: 4.6 % / Biso Wilson estimate: 23.7 Å2 / Rsym value: 0.045 / Net I/σ(I): 12.5 |
| Reflection shell | Resolution: 1.82→1.87 Å / Redundancy: 4.5 % / Mean I/σ(I) obs: 3.4 / Rsym value: 0.25 / % possible all: 57 |
| Reflection | *PLUS Rmerge(I) obs: 0.045 |
| Reflection shell | *PLUS % possible obs: 57 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1KIM Resolution: 1.9→16 Å / Isotropic thermal model: INDIVIDUAL B-FACTOR REFINEMENT / Cross valid method: FREE R / σ(F): 3 / Details: NCS RESTRAINTS NOT USED IN FINAL CYCLES.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→16 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: NCS RESTRAINT NOT USED IN FINAL REFT. STAGES. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.97 Å / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / % reflection Rfree: 10 % / Rfactor obs: 0.241 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.9 Å |
