| 登録情報 | データベース: PDB / ID: 2hl0
|
|---|
| タイトル | Crystal structure of the editing domain of threonyl-tRNA synthetase from Pyrococcus abyssi in complex with seryl-3'-aminoadenosine |
|---|
 要素 要素 | Threonyl-tRNA synthetase |
|---|
 キーワード キーワード |  LIGASE (リガーゼ) / translation (翻訳 (生物学)) / editing (編集) / aminoacyl-tRNA synthetase (アミノアシルtRNA合成酵素) / enzyme mechanism (酵素反応) / enantioselectivity (光学異性体) LIGASE (リガーゼ) / translation (翻訳 (生物学)) / editing (編集) / aminoacyl-tRNA synthetase (アミノアシルtRNA合成酵素) / enzyme mechanism (酵素反応) / enantioselectivity (光学異性体) |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
トレオニンtRNAリガーゼ / threonyl-tRNA aminoacylation / threonine-tRNA ligase activity / tRNA binding / zinc ion binding / ATP binding / 細胞質基質類似検索 - 分子機能 Threonyl-tRNA synthetase, editing domain, archaea / Archaea-specific editing domain of threonyl-tRNA synthetase / D-tyrosyl-tRNA(Tyr) deacylase / D-aminoacyl-tRNA deacylase-like superfamily / Threonine-tRNA ligase, class IIa / Threonine-tRNA ligase catalytic core domain / : / D-tyrosyl-trna(Tyr) Deacylase; Chain: A; / Aminoacyl-tRNA synthetase, class II (G/ P/ S/T) / tRNA synthetase class II core domain (G, H, P, S and T) ...Threonyl-tRNA synthetase, editing domain, archaea / Archaea-specific editing domain of threonyl-tRNA synthetase / D-tyrosyl-tRNA(Tyr) deacylase / D-aminoacyl-tRNA deacylase-like superfamily / Threonine-tRNA ligase, class IIa / Threonine-tRNA ligase catalytic core domain / : / D-tyrosyl-trna(Tyr) Deacylase; Chain: A; / Aminoacyl-tRNA synthetase, class II (G/ P/ S/T) / tRNA synthetase class II core domain (G, H, P, S and T) / Anticodon-binding / Anticodon binding domain / Anticodon-binding domain superfamily / Aminoacyl-tRNA synthetase, class II / Aminoacyl-transfer RNA synthetases class-II family profile. / Class II Aminoacyl-tRNA synthetase/Biotinyl protein ligase (BPL) and lipoyl protein ligase (LPL) / 3-Layer(bba) Sandwich / Alpha Beta類似検索 - ドメイン・相同性 SERINE-3'-AMINOADENOSINE / Threonine--tRNA ligase類似検索 - 構成要素 |
|---|
| 生物種 |   Pyrococcus abyssi (古細菌) Pyrococcus abyssi (古細菌) |
|---|
| 手法 | X線回折 / 分子置換 / 解像度: 1.86 Å |
|---|
 データ登録者 データ登録者 | Hussain, T. / Kruparani, S.P. / Pal, B. / Sankaranarayanan, R. |
|---|
 引用 引用 | |
|---|
| 履歴 | | 登録 | 2006年7月6日 | 登録サイト: RCSB / 処理サイト: PDBJ |
|---|
| 改定 1.0 | 2006年8月29日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2008年5月1日 | Group: Version format compliance |
|---|
| 改定 1.2 | 2011年7月13日 | Group: Version format compliance |
|---|
| 改定 1.3 | 2023年10月25日 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード













 PDBj
PDBj
 集合体
集合体


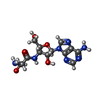
 分子量: 353.334 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C13H19N7O5
分子量: 353.334 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C13H19N7O5 分子量: 18.015 Da / 分子数: 199 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 199 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析