 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2c6u | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human CLEC-2 (CLEC1B) | ||||||
 Components Components | CLEC1B PROTEIN | ||||||
 Keywords Keywords |  LECTIN / CLEC-2 / RHODOCYTIN / AGGRETIN / C-TYPE LECTIN-LIKE / PLATELETS / THROMBOSIS / CLEC1B LECTIN / CLEC-2 / RHODOCYTIN / AGGRETIN / C-TYPE LECTIN-LIKE / PLATELETS / THROMBOSIS / CLEC1B | ||||||
| Function / homology |  Function and homology information Function and homology informationplatelet formation / GPVI-mediated activation cascade / Heme signaling / defense response / transmembrane signaling receptor activity / carbohydrate binding / cell surface receptor signaling pathway / cell surface / signal transduction / plasma membraneSimilarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Watson, A.A. / Brown, J. / O'Callaghan, C.A. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2007 Title: The Crystal Structure and Mutational Binding Analysis of the Extracellular Domain of the Platelet-Activating Receptor Clec-2. Authors: Watson, A.A. / Brown, J. / Harlos, K. / Eble, J.A. / Walter, T.S. / O'Callaghan, C.A. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
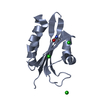
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2c6u.cif.gz | 45.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2c6u.ent.gz | 31.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2c6u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c6/2c6uftp://data.pdbj.org/pub/pdb/validation_reports/c6/2c6u | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14601.466 Da / Num. of mol.: 1 / Fragment: C-TYPE LECTIN-LIKE DOMAIN, RESIDUES 100-221 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Cell: PERIPHERAL BLOOD MONONUCLEAR CELLS / Plasmid: PGMT7 / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): PLYSS / References: UniProt: Q9P126 ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / Variant (production host): PLYSS / References: UniProt: Q9P126 |
|---|---|
| #2: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 246 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 246 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | MODELLED AS GLY, PROVEN BY DNA SEQUENCING AND MASS SPECTROSCOPY TO BE SER UNIPROT REFERENCE Q8NHR6_ ...MODELLED AS GLY, PROVEN BY DNA SEQUENCING |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.81 Å3/Da / Density % sol: 31.99 % Description: A COMPOSITE MODEL WAS CONSTRUCTED USING THE CASPR WEBSERVER FROM THE FOLLOWING PDB ENTRIES 1MPU, 1K9I, 1P4L, 1QO3, 1XPH AND 1YPQ. |
|---|---|
| Crystal grow | pH: 6.5 Details: 20% W/V POLYETHYLENE GLYCOL MONOMETHYL ETHER 5000 0.1M BIS-TRIS PH 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX14.1 / Wavelength: 1.488 / Beamline: PX14.1 / Wavelength: 1.488 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Oct 28, 2005 / Details: MIRRORS |
| Radiation | Monochromator: ASYMMETRIC CUT SI (111) CRYTALS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.488 Å / Relative weight: 1 |
| Reflection | Resolution: 1.44→50 Å / Num. obs: 20396 / % possible obs: 88 % / Observed criterion σ(I): -3 / Redundancy: 11.4 % / Rmerge(I) obs: 0.04 / Net I/σ(I): 49.02 |
| Reflection shell | Resolution: 1.44→1.49 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.11 / Mean I/σ(I) obs: 10.13 / % possible all: 24.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: COMPOSITE MODEL - SEE REMARKS Resolution: 1.6→39.31 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.945 / SU B: 1.593 / SU ML: 0.058 / Cross valid method: THROUGHOUT / ESU R: 0.1 / ESU R Free: 0.099 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13.02 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→39.31 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|