 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2c28: Efficient and High Fidelity Incorporation of dCTP Opposite 7,8- D... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2c28 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Efficient and High Fidelity Incorporation of dCTP Opposite 7,8- Dihydro-8-oxodeoxyguanosine by Sulfolobus solfataricus DNA Polymerase Dpo4 | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  POLYMERASE / 7 / 8-DIHYDRO-8-OXODEOXYGUANOSINE / P2 DNA POLYMERASE IV / TRANSLESION DNA POLYMERASE / DDGTP / NUCLEOTIDYLTRANSFERASE / TRANSFERASE / DNA DAMAGE / DNA REPAIR / DNA REPLICATION / DNA- BINDING / DNA-DIRECTED DNA POLYMERASE / MAGNESIUM / METAL- BINDING / MUTATOR PROTEIN POLYMERASE / 7 / 8-DIHYDRO-8-OXODEOXYGUANOSINE / P2 DNA POLYMERASE IV / TRANSLESION DNA POLYMERASE / DDGTP / NUCLEOTIDYLTRANSFERASE / TRANSFERASE / DNA DAMAGE / DNA REPAIR / DNA REPLICATION / DNA- BINDING / DNA-DIRECTED DNA POLYMERASE / MAGNESIUM / METAL- BINDING / MUTATOR PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationerror-prone translesion synthesis / DNA-templated DNA replication / damaged DNA binding / DNA-directed DNA polymerase / DNA-directed DNA polymerase activity / magnesium ion binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |   SULFOLOBUS SOLFATARICUS (archaea) SULFOLOBUS SOLFATARICUS (archaea) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.27 Å | ||||||
 Authors Authors | Irimia, A. / Loukachevitch, L.V. / Egli, M. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2006 Title: Efficient and High Fidelity Incorporation of Dctp Opposite 7,8-Dihydro-8-Oxodeoxyguanosine by Sulfolobus Solfataricus DNA Polymerase Dpo4 Authors: Zang, H. / Irimia, A. / Choi, J.-Y. / Angel, K.C. / Loukachevitch, L.V. / Egli, M. / P Guengerich, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2c28.cif.gz | 103.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2c28.ent.gz | 75.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2c28.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c2/2c28ftp://data.pdbj.org/pub/pdb/validation_reports/c2/2c28 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2c22C  2c2dC  2c2eC  2c2rC  2br0S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




























-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | / POL IV Mass: 41086.754 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) SULFOLOBUS SOLFATARICUS (archaea) / Strain: P2 / Description: GENE DPO4 / Plasmid: PET22B/DPO4-NHIS / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q97W02, DNA-directed DNA polymerase ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q97W02, DNA-directed DNA polymerase |
|---|
-DNA chain , 2 types, 2 molecules PT
| #2: DNA chain | Mass: 4096.670 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: 13-MER PRIMER |
|---|---|
| #3: DNA chain | Mass: 5388.480 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: 18-MER TEMPLATE CONTAINING A 7,8-DIHYDRO-8-OXODEOXYGUANOSINE |
-Non-polymers , 3 types, 102 molecules 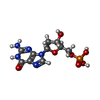


| #4: Chemical | ChemComp-DG / Deoxyguanosine monophosphate Type: DNA linking / Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: dGMP*YM Type: DNA linking / Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: dGMP*YM | ||
|---|---|---|---|
| #5: Chemical |  Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Compound details | INVOLVEMEN |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 50.96 % Description: THE STARTING MODEL WAS THE PDB FILE WITH THE ACCESSION CODE 2BR0. ITS POSITION WAS OPTIMIZED BY SEVERAL ROUNDS OF RIGID BODY REFINEMENT |
|---|---|
| Crystal grow | pH: 7.5 Details: 5% PEG 3350,0.010 M TRIS-HCL PH 7.5, 100 MM CA(CH3CO2)2, 1.25% GLYCEROL, 30 MM NACL |
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 5ID-B / Wavelength: 1 / Beamline: 5ID-B / Wavelength: 1 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Aug 1, 2005 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.27→31.96 Å / Num. obs: 23827 / % possible obs: 99.7 % / Observed criterion σ(I): 1 / Redundancy: 7.08 % / Biso Wilson estimate: 56.2 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 10.41 |
| Reflection shell | Resolution: 2.27→2.42 Å / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2BR0 Resolution: 2.27→31.96 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 1204106.46 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 43.7616 Å2 / ksol: 0.331059 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.27→31.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.27→2.42 Å / Rfactor Rfree error: 0.026 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
