 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2bv3: Crystal structure of a mutant elongation factor G trapped with a ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2bv3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a mutant elongation factor G trapped with a GTP analogue | ||||||
 Components Components | ELONGATION FACTOR G EF-G EF-G | ||||||
 Keywords Keywords | ELONGATION FACTOR / SWITCH II / GTP-BINDING / TRANSLATION MUTATION THR84ALA / PROTEIN BIOSYNTHESIS | ||||||
| Function / homology |  Function and homology information Function and homology informationtranslational elongation / translation elongation factor activity / GDP binding / ribosome binding / GTPase activity / GTP binding / magnesium ion binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |   THERMUS THERMOPHILUS (bacteria) THERMUS THERMOPHILUS (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Hansson, S. / Singh, R. / Gudkov, A.T. / Liljas, A. / Logan, D.T. | ||||||
 Citation Citation | Journal: FEBS Lett. / Year: 2005 Title: Crystal Structure of a Mutant Elongation Factor G Trapped with a GTP Analogue. Authors: Hansson, S. / Singh, R. / Gudkov, A.T. / Liljas, A. / Logan, D.T. #1: Journal: Embo J. / Year: 1994Title: The Crystal Structure of Elongation Factor G Complexed with Gdp, at 2.7 A Resolution Authors: Czworkowski, J. / Wang, J. / Steitz, T.A. / Moore, P.B. #2: Journal: EMBO J / Year: 1994Title: Three-dimensional structure of the ribosomal translocase: elongation factor G from Thermus thermophilus. Authors: A AEvarsson / E Brazhnikov / M Garber / J Zheltonosova / Y Chirgadze / S al-Karadaghi / L A Svensson / A Liljas /  Abstract: The crystal structure of Thermus thermophilus elongation factor G without guanine nucleotide was determined to 2.85 A. This GTPase has five domains with overall dimensions of 50 x 60 x 118 A. The GTP ...The crystal structure of Thermus thermophilus elongation factor G without guanine nucleotide was determined to 2.85 A. This GTPase has five domains with overall dimensions of 50 x 60 x 118 A. The GTP binding domain has a core common to other GTPases with a unique subdomain which probably functions as an intrinsic nucleotide exchange factor. Domains I and II are homologous to elongation factor Tu and their arrangement, both with and without GDP, is more similar to elongation factor Tu in complex with a GTP analogue than with GDP. Domains III and V show structural similarities to ribosomal proteins. Domain IV protrudes from the main body of the protein and has an extraordinary topology with a left-handed cross-over connection between two parallel beta-strands. #3: Journal: Structure / Year: 1996Title: The Structure of Elongation Factor G in Complex with Gdp: Conformational Flexibility and Nucleotide Exchange Authors: Al-Karadaghi, S. / Aevarsson, A. / Garber, M. / Zheltonosova, J. / Liljas, A. #4: Journal: J.Mol.Biol. / Year: 2000Title: Structure of a Mutant EF-G Reveals Domain III and Possibly the Fusidic Acid Binding Site Authors: Laurberg, M. / Kristensen, O. / Martemyanov, K. / Gudkov, A.T. / Nagaev, I. / Hughes, D. / Liljas, A. #5: Journal: J.Biol.Chem. / Year: 2001 Title: Mutations in the G-Domain of Elongation Factor G from Thermus Thermophilus Affect Both its Interaction with GTP and Fusidic Acid. Authors: Martemyanov, K.A. / Liljas, A. / Yarunin, A.S. / Gudkov, A.T. #6: Journal: J.Mol.Biol. / Year: 2005Title: Structural Insights Into Fusidic Acid Resistance and Sensitivity in EF-G Authors: Hansson, S. / Singh, R. / Gudkov, A.T. / Liljas, A. / Logan, D.T. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bv3.cif.gz | 142.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bv3.ent.gz | 109.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2bv3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bv/2bv3ftp://data.pdbj.org/pub/pdb/validation_reports/bv/2bv3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2bm0S S: Starting model for refinement |
|---|---|
| Similar structure data |
























-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | EF-G / EF-G Mass: 76756.836 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) THERMUS THERMOPHILUS (bacteria) / Plasmid: PET13A / Production host: ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P13551 |
|---|---|
| #2: Chemical | ChemComp-GNP / 5'-Guanylyl imidodiphosphate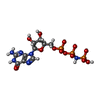  Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3 Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3Comment: GppNHp, GMPPNP, energy-carrying molecule analogue*YM |
| #3: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | ENGINEERED |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3 Å3/Da / Density % sol: 58.6 % |
|---|---|
| Crystal grow | pH: 7.3 Details: 17 % PEG8000, 10 MM MGCL2, 100 MM GDPNP, 46 MM TRIS, 100 MM HEPES, pH 7.30 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II / Beamline: I711 / Wavelength: 0.9787 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Apr 16, 2004 / Details: MIRROR |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9787 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→26.9 Å / Num. obs: 30656 / % possible obs: 96.5 % / Observed criterion σ(I): 2.5 / Redundancy: 7.6 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 11.5 |
| Reflection shell | Resolution: 2.5→2.6 Å / Redundancy: 6.5 % / Rmerge(I) obs: 0.44 / Mean I/σ(I) obs: 2.7 / % possible all: 94.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2BM0 Resolution: 2.5→17.43 Å / Cor.coef. Fo:Fc: 0.948 / Cor.coef. Fo:Fc free: 0.905 / SU B: 25.057 / SU ML: 0.246 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.394 / ESU R Free: 0.292 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. RESIDUES 1-6, 42-60, 689-691 ARE MISSING.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53.29 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→17.43 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|