- PDB-2bo1: Crystal structure of a hybrid ribosomal protein L30e with surface... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 2bo1
Title
Crystal structure of a hybrid ribosomal protein L30e with surface residues from T. celer, and core residues from yeast
Components
50S RIBOSOMAL PROTEIN L30ERibosome
Keywords
RIBOSOMAL PROTEIN / RIBONUCLEOPROTEIN
Function / homology
Function and homology information
cytosolic large ribosomal subunit / structural constituent of ribosome / translation / RNA binding Similarity search - Function
Ribosomal protein L30/S12 / Ribosomal protein L30e / Ribosomal protein L30e, conserved site / Ribosomal protein L30/YlxQ / 60s Ribosomal Protein L30; Chain: A; / Ribosomal protein L30e signature 1. / Ribosomal protein L30e signature 2. / Ribosomal protein L7Ae/L30e/S12e/Gadd45 / Ribosomal protein L7Ae/L30e/S12e/Gadd45 family / 50S ribosomal protein L30e-like ...Ribosomal protein L30/S12 / Ribosomal protein L30e / Ribosomal protein L30e, conserved site / Ribosomal protein L30/YlxQ / 60s Ribosomal Protein L30; Chain: A; / Ribosomal protein L30e signature 1. / Ribosomal protein L30e signature 2. / Ribosomal protein L7Ae/L30e/S12e/Gadd45 / Ribosomal protein L7Ae/L30e/S12e/Gadd45 family / 50S ribosomal protein L30e-like / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: H2O
Compound details
ENGINEERED RESIDUE IN CHAIN A, PHE 4 TO ILE ENGINEERED RESIDUE IN CHAIN A, ALA 11 TO VAL ENGINEERED ...ENGINEERED RESIDUE IN CHAIN A, PHE 4 TO ILE ENGINEERED RESIDUE IN CHAIN A, ALA 11 TO VAL ENGINEERED RESIDUE IN CHAIN A, GLN 12 TO ILE ENGINEERED RESIDUE IN CHAIN A, THR 14 TO SER ENGINEERED RESIDUE IN CHAIN A, ILE 17 TO TYR ENGINEERED RESIDUE IN CHAIN A, VAL 18 TO THR ENGINEERED RESIDUE IN CHAIN A, MET 19 TO LEU ENGINEERED RESIDUE IN CHAIN A, ALA 21 TO TYR ENGINEERED RESIDUE IN CHAIN A, SER 24 TO THR ENGINEERED RESIDUE IN CHAIN A, ILE 25 TO VAL ENGINEERED RESIDUE IN CHAIN A, TYR 27 TO SER ENGINEERED RESIDUE IN CHAIN A, ALA 28 TO LEU ENGINEERED RESIDUE IN CHAIN A, ALA 33 TO SER ENGINEERED RESIDUE IN CHAIN A, VAL 38 TO ILE ENGINEERED RESIDUE IN CHAIN A, ALA 42 TO THR ENGINEERED RESIDUE IN CHAIN A, ILE 46 TO ARG ENGINEERED RESIDUE IN CHAIN A, ILE 50 TO LEU ENGINEERED RESIDUE IN CHAIN A, ILE 59 TO THR ENGINEERED RESIDUE IN CHAIN A, SER 68 TO ASN ENGINEERED RESIDUE IN CHAIN A, LEU 74 TO ALA ENGINEERED RESIDUE IN CHAIN A, LEU 75 TO VAL ENGINEERED RESIDUE IN CHAIN A, ARG 77 TO LYS ENGINEERED RESIDUE IN CHAIN A, ALA 83 TO VAL ENGINEERED RESIDUE IN CHAIN A, LEU 84 TO VAL ENGINEERED RESIDUE IN CHAIN A, ALA 85 TO SER ENGINEERED RESIDUE IN CHAIN A, VAL 86 TO ILE ENGINEERED RESIDUE IN CHAIN A, VAL 87 TO LEU ENGINEERED RESIDUE IN CHAIN A, PRO 89 TO ALA
Sequence details
THE AUTHORS SAYS THE ENTRY CAN BE CONSIDERED AS A MUTANT OF T. CELER L30E WITH THE FOLLOWING MUTATIONS
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 1.58 Å3/Da / Density % sol: 21.46 %
Crystal grow
pH: 8.5 Details: 2UL 10MG/ML PROTEIN AND 2UL 22.5% PEG3350, 0.1M TRIS PH 8.5, CRYOPROTECTED IN 20% PEG 400
Resolution: 1.7→37.19 Å / Cor.coef. Fo:Fc: 0.938 / Cor.coef. Fo:Fc free: 0.85 / SU B: 1.954 / SU ML: 0.069 / Cross valid method: THROUGHOUT / ESU R: 0.127 / ESU R Free: 0.135 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.24
445
4.8 %
RANDOM
Rwork
0.173
-
-
-
obs
0.176
8796
98.5 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Ribosome
Ribosome  Keywords
Keywords Function and homology information
Function and homology information

 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






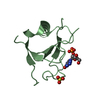







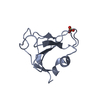
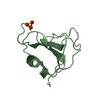
 PDBj
PDBj




 Assembly
Assembly


 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 6B / Wavelength: 1.12714
/ Beamline: 6B / Wavelength: 1.12714  Processing
Processing