 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1zfj: INOSINE MONOPHOSPHATE DEHYDROGENASE (IMPDH; EC 1.1.1.205) FROM ST... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1zfj | ||||||
|---|---|---|---|---|---|---|---|
| Title | INOSINE MONOPHOSPHATE DEHYDROGENASE (IMPDH; EC 1.1.1.205) FROM STREPTOCOCCUS PYOGENES | ||||||
 Components Components | INOSINE MONOPHOSPHATE DEHYDROGENASE | ||||||
 Keywords Keywords |  OXIDOREDUCTASE / IMPDH / DEHYDROGENASE / CBS DOMAINS OXIDOREDUCTASE / IMPDH / DEHYDROGENASE / CBS DOMAINS | ||||||
| Function / homology |  Function and homology informationIMP dehydrogenase activity / IMP dehydrogenase / GMP biosynthetic process / nucleotide binding / metal ion binding Function and homology informationIMP dehydrogenase activity / IMP dehydrogenase / GMP biosynthetic process / nucleotide binding / metal ion bindingSimilarity search - Function | ||||||
| Biological species |  Streptococcus pyogenes (bacteria) Streptococcus pyogenes (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.9 Å | ||||||
 Authors Authors | Zhang, R. / Evans, G. / Rotella, F.J. / Westbrook, E.M. / Beno, D. / Huberman, E. / Joachimiak, A. / Collart, F.R. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: Characteristics and crystal structure of bacterial inosine-5'-monophosphate dehydrogenase. Authors: Zhang, R. / Evans, G. / Rotella, F.J. / Westbrook, E.M. / Beno, D. / Huberman, E. / Joachimiak, A. / Collart, F.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1zfj.cif.gz | 117 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1zfj.ent.gz | 89 KB | Display | PDB format |
| PDBx/mmJSON format | 1zfj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zf/1zfjftp://data.pdbj.org/pub/pdb/validation_reports/zf/1zfj | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|







-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | x 8
| ||||||||
| Unit cell |
| ||||||||
| Details | S. PYOGENES IMPDH IS A TETRAMER WITH ITS FOUR SUBUNITS RELATED BY A CRYSTALLOGRAPHIC FOURFOLD AXIS. EACH MONOMER HAS A TWO-DOMAIN STRUCTURE: A CATALYTIC DOMAIN (AMINO ACID RESIDUES 2-92 AND 224-492) FORMING THE INTERIOR CORE OF THE ACTIVE TETRAMERIC ENZYME AND A CBS DIMER DOMAIN (RESIDUES 93-223) PROJECTING OUTWARD FROM THE CORNERS OF THE SQUARE. THE CBS DESIGNATION ARISES FROM THE ORIGINAL IDENTIFICATION OF THIS FOLDING MOTIF IN THE ENZYME CYSTATHIONINE-"BETA"-SYNTHASE [BATEMAN, A. (1997) TRENDS BIOCHEM. SCI. 22, 12-13]. THE CBS DIMER DOMAIN, FOUND IN IMPDH PROTEINS FROM ALL THREE KINGDOMS, IS COMPOSED OF TWO CBS MOTIFS RELATED BY APPROXIMATE TWOFOLD SYMMETRY (RMS DEVIATIONS BETWEEN ALPHA CARBON ATOMS: 2.7 ANGSTROMS). EACH CBS MOTIF HAS THE CHARACTERISTIC SHEET/HELIX/SHEET/SHEET/HELIX TOPOLOGY. THIS IS THE FIRST REPORTED COMPLETE STRUCTURE OF A CBS DIMER DOMAIN, A FOLDING MOTIF PROPOSED TO ACT AS A REGULATORY ELEMENT SINCE MUTATIONS LEAD TO THE HUMAN DISEASE HOMOCYSTINURIA. EACH IPMDH MONOMER CONTAINS IMP IN THE CATALYTIC SITE. THIS SUBSTRATE IS NOT COVALENTLY BOUND TO THE ACTIVE SITE CYS310 SUGGESTING THAT IMP DOES NOT FORM A COVALENT BOND IN THE ABSENCE OF NAD. |
-Components
| #1: Protein | Mass: 53228.352 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN, CBS DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus pyogenes (bacteria) / Production host: Streptococcus pyogenes (bacteria) / Strain (production host): ESCHERICHIA COLI / References: UniProt: P0C0H6, IMP dehydrogenase |
|---|---|
| #2: Chemical | ChemComp-IMP / Inosinic acid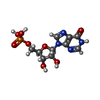  Mass: 348.206 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N4O8P Mass: 348.206 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N4O8P |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 499 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 499 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.74 Å3/Da / Density % sol: 49 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.2 Details: 0.1 M MES (PH 7.2), 1.8 M AMMONIUM SULFATE, 10 MM COCL2, pH 7.20 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 10 ℃ / pH: 7.4 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.9791,1.0781 / Beamline: 19-ID / Wavelength: 0.9791,1.0781 | |||||||||
| Detector | Type: SBC-1 / Detector: CCD / Details: MIRROR | |||||||||
| Radiation | Monochromator: SI(111) / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 1.9→40 Å / Num. obs: 44921 / % possible obs: 96.5 % / Observed criterion σ(I): 0 / Redundancy: 6.2 % / Biso Wilson estimate: 21.9 Å2 / Rmerge(I) obs: 0.068 / Net I/σ(I): 6 | |||||||||
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 3 % / Rmerge(I) obs: 0.319 / Mean I/σ(I) obs: 2.5 / % possible all: 87.6 | |||||||||
| Reflection | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 40 Å / % possible obs: 96.5 % / Observed criterion σ(I): 0 / Redundancy: 6.2 % / Num. measured all: 263355 | |||||||||
| Reflection shell | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 1.97 Å / Redundancy: 3 % / Mean I/σ(I) obs: 2.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.9→6 Å / Rfactor Rfree error: 0.004 / Data cutoff high rms absF: 986591.3 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→2.01 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.3 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 6 Å / Rfactor obs: 0.234 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.368 / Rfactor Rwork: 0.357 |
