 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ymg: The Channel Architecture of Aquaporin O at 2.2 Angstrom Resolution -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ymg | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | The Channel Architecture of Aquaporin O at 2.2 Angstrom Resolution | ||||||||||||
 Components Components | Lens fiber major intrinsic protein | ||||||||||||
 Keywords Keywords | MEMBRANE PROTEIN / AQP0 / Integral Membrane Protein / MIP26 / Lens / Cataract / Water Channel | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationPassive transport by Aquaporins / gap junction-mediated intercellular transport / water channel activity / water transport / structural constituent of eye lens / gap junction / lens development in camera-type eye / positive regulation of cell adhesion / visual perception / protein homotetramerization ...Passive transport by Aquaporins / gap junction-mediated intercellular transport / water channel activity / water transport / structural constituent of eye lens / gap junction / lens development in camera-type eye / positive regulation of cell adhesion / visual perception / protein homotetramerization / calmodulin binding / apical plasma membrane / endoplasmic reticulum / plasma membraneSimilarity search - Function | ||||||||||||
| Biological species |  Bos taurus (cattle) Bos taurus (cattle) | ||||||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.24 Å | ||||||||||||
 Authors Authors | Harries, W.E.C. / Akhavan, D. / Miercke, L.J.W. / Khademi, S. / Stroud, R.M. | ||||||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2004 Title: The Channel Architecture of Aquaporin 0 at a 2.2-A Resolution Authors: Harries, W.E.C. / Akhavan, D. / Miercke, L.J.W. / Khademi, S. / Stroud, R.M. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ymg.cif.gz | 62.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ymg.ent.gz | 44.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1ymg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ym/1ymgftp://data.pdbj.org/pub/pdb/validation_reports/ym/1ymg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1j4nS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | The monomers arrange into a tetramer generated from the monomer by the operations: -x,-y,z: 1/2-y, 1/2: 1/2+y,1/2 |
-Components
| #1: Protein | / MIP26 / MP26 / AQP0 Mass: 28244.865 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: Ocular lens fiber cell plasma membrane / Source: (natural) Bos taurus (cattle) / Cell: FIBER / Cellular location: PLASMA MEMBRANECell membrane / Organ: EYE / Tissue: LENS / References: UniProt: P06624 | ||
|---|---|---|---|
| #2: Sugar | 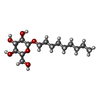  Type: D-saccharide / Mass: 306.395 Da / Num. of mol.: 2 Type: D-saccharide / Mass: 306.395 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C15H30O6 / Comment: detergent*YM #3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 182 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 182 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.89 Å3/Da / Density % sol: 55.78 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 10 Details: 30% polyethylene glycol 1000, 20 mM glycine, 50 mM NaCl, pH 10.00, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.3.1 / Wavelength: 1.11587 / Wavelength: 1.11587 Å / Beamline: 8.3.1 / Wavelength: 1.11587 / Wavelength: 1.11587 Å | |||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Oct 15, 2003 | |||||||||
| Radiation | Monochromator: DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 2.2→30 Å / Num. all: 16385 / Num. obs: 16284 / % possible obs: 93.7 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 20.3 % / Biso Wilson estimate: 25.7 Å2 / Rmerge(I) obs: 0.041 / Rsym value: 0.041 / Net I/σ(I): 27.96 | |||||||||
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 5.3 % / Rmerge(I) obs: 0.5 / Mean I/σ(I) obs: 2.6 / Rsym value: 0.5 / % possible all: 80 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1J4N Resolution: 2.24→25.26 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 328368.91 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 93.2556 Å2 / ksol: 0.327246 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53.1 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.24→25.26 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.34 Å / Rfactor Rfree error: 0.041 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
