Mass: 18.015 Da / Num. of mol.: 1071 / Source method: isolated from a natural source / Formula: H2O
Compound details
ENZYME OF PLANT METABOLISM CATALYZING THE FIRST REACTION IN THE BIOSYNTHESIS FROM L-PHENYLALANINE. ...ENZYME OF PLANT METABOLISM CATALYZING THE FIRST REACTION IN THE BIOSYNTHESIS FROM L-PHENYLALANINE. CONTAINS AN ACTIVE SITE 4-METHYLIDENE-IMIDAZOLE-5-ONE (MIO), WHICH IS FORMED AUTOCATALYTICALLY BY CYCLIZATION AND DEHYDRATION OF RESIDUES ALA-SER-GLY.
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components
 Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads





 PDBj
PDBj




 Assembly
Assembly


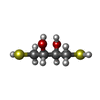
 Mass: 154.251 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O2S2
Mass: 154.251 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O2S2 Mass: 18.015 Da / Num. of mol.: 1071 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 1071 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: PX9.5 / Wavelength: 0.9798,0.9800,0.9824
/ Beamline: PX9.5 / Wavelength: 0.9798,0.9800,0.9824 Processing
Processing