 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1vtk: THYMIDINE KINASE FROM HERPES SIMPLEX VIRUS TYPE 1 IN COMPLEX WITH... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1vtk | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | THYMIDINE KINASE FROM HERPES SIMPLEX VIRUS TYPE 1 IN COMPLEX WITH ADP AND DEOXYTHYMIDINE-MONOPHOSPHATE | |||||||||
 Components Components | THYMIDINE KINASE | |||||||||
 Keywords Keywords | TRANSFERASE / KEY ENZYME IN THYMIDINE SALVAGE PATHWAY / ADDITIONAL THYMIDYLATE KINASE ACTIVITY / TARGET FOR ANTI-HERPES VIRAL DRUGS | |||||||||
| Function / homology |  Function and homology information Function and homology informationTMP biosynthetic process / thymidine kinase / thymidine kinase activity / DNA biosynthetic process / phosphorylation / ATP bindingSimilarity search - Function | |||||||||
| Biological species |  Herpes simplex virus Herpes simplex virus | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 2.75 Å | |||||||||
 Authors Authors | Wild, K. / Schulz, G.E. | |||||||||
 Citation Citation | Journal: Protein Sci. / Year: 1997 Title: The structures of thymidine kinase from herpes simplex virus type 1 in complex with substrates and a substrate analogue. Authors: Wild, K. / Bohner, T. / Folkers, G. / Schulz, G.E. #1: Journal: FEBS Lett. / Year: 1995Title: The Three-Dimensional Structure of Thymidine Kinase from Herpes Simplex Virus Type 1 Authors: Wild, K. / Bohner, T. / Aubry, A. / Folkers, G. / Schulz, G.E. #2: Journal: Protein Expr.Purif. / Year: 1994Title: A Fast Method for Obtaining Highly Pure Recombinant Herpes Simplex Virus Type 1 Thymidine Kinase Authors: Fetzer, J. / Michael, M. / Bohner, T. / Hofbauer, R. / Folkers, G. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1vtk.cif.gz | 76 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1vtk.ent.gz | 55.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1vtk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vt/1vtkftp://data.pdbj.org/pub/pdb/validation_reports/vt/1vtk | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 37205.605 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Herpes simplex virus (type 1 / strain F)Genus: Simplexvirus / Species: Human herpesvirus 1Herpesviridae / Strain: F / Plasmid: PGEX2T / Production host:  Escherichia coli (E. coli) / Strain (production host): KY 895 Escherichia coli (E. coli) / Strain (production host): KY 895References: UniProt: P03176, UniProt: P0DTH5*PLUS, thymidine kinase |
|---|---|
| #2: Chemical | ChemComp-ADP / Adenosine diphosphate  Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM |
| #3: Chemical | ChemComp-TMP / 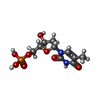  Mass: 322.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O8P Mass: 322.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O8P |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 52 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 52 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.65 Å3/Da / Density % sol: 67 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.5 / Details: pH 5.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop / Details: Wild, K., (1995) FEBS Lett., 368, 289. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X31 / Wavelength: 0.9204 / Beamline: X31 / Wavelength: 0.9204 |
| Detector | Date: Mar 1, 1995 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9204 Å / Relative weight: 1 |
| Reflection | Resolution: 2.75→10 Å / Num. obs: 12164 / % possible obs: 89.5 % / Redundancy: 2.3 % / Biso Wilson estimate: 31 Å2 / Rsym value: 0.092 / Net I/σ(I): 17.7 |
| Reflection shell | Resolution: 2.75→2.8 Å / Redundancy: 2.2 % / Mean I/σ(I) obs: 5.7 / Rsym value: 0.412 / % possible all: 91.2 |
| Reflection | *PLUS Num. measured all: 28046 / Rmerge(I) obs: 0.092 |
| Reflection shell | *PLUS % possible obs: 91.2 % / Rmerge(I) obs: 0.412 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 2.75→10 Å / Rfactor Rfree error: 0.007 / Cross valid method: THROUGHOUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.27 Å / Luzzati d res low obs: 4.8 Å / Luzzati sigma a obs: 0.46 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.75→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.75→2.87 Å / Rfactor Rfree error: 0.028 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
