 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1uob: Deacetoxycephalosporin C synthase complexed with 2-oxoglutarate a... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1uob | ||||||
|---|---|---|---|---|---|---|---|
| Title | Deacetoxycephalosporin C synthase complexed with 2-oxoglutarate and penicillin G | ||||||
 Components Components | DEACETOXYCEPHALOSPORIN C SYNTHETASE | ||||||
 Keywords Keywords |  OXIDOREDUCTASE / ANTIBIOTIC BIOSYNTHESIS OXIDOREDUCTASE / ANTIBIOTIC BIOSYNTHESIS | ||||||
| Function / homology |  Function and homology informationdeacetoxycephalosporin-C synthase / deacetoxycephalosporin-C synthase activity / L-ascorbic acid binding / antibiotic biosynthetic process / iron ion binding Function and homology informationdeacetoxycephalosporin-C synthase / deacetoxycephalosporin-C synthase activity / L-ascorbic acid binding / antibiotic biosynthetic process / iron ion bindingSimilarity search - Function | ||||||
| Biological species |  STREPTOMYCES CLAVULIGERUS (bacteria) STREPTOMYCES CLAVULIGERUS (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Valegard, K. / Terwisscha van Scheltinga, A.C. / Dubus, A. / Oster, L.M. / Rhangino, G. / Hajdu, J. / Andersson, I. | ||||||
 Citation Citation | Journal: Nat.Struct.Mol.Biol. / Year: 2004 Title: The Structural Basis of Cephalosporin Formation in a Mononuclear Ferrous Enzyme Authors: Valegard, K. / Terwisscha van Scheltinga, A.C. / Dubus, A. / Ranghino, G. / Oster, L.M. / Hajdu, J. / Andersson, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1uob.cif.gz | 76.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1uob.ent.gz | 55.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1uob.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/uo/1uobftp://data.pdbj.org/pub/pdb/validation_reports/uo/1uob | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1unbC  1uo9C  1uofC  1uogC  1rxfS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 34591.641 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) STREPTOMYCES CLAVULIGERUS (bacteria) / Plasmid: PET11A / Production host: ESCHERICHIA COLI (E. coli)References: UniProt: P18548, deacetoxycephalosporin-C synthase |
|---|---|
| #2: Chemical | ChemComp-FE2 /   Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe |
| #3: Chemical | ChemComp-PNN / Benzylpenicillin  Mass: 334.390 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H18N2O4S / Comment: antibiotic*YM Mass: 334.390 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H18N2O4S / Comment: antibiotic*YM |
| #4: Chemical | ChemComp-AKG / Α-Ketoglutaric acid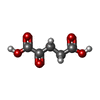  Mass: 146.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H6O5 Mass: 146.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H6O5 |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 156 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 156 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.26 Å3/Da / Density % sol: 45.65 % Description: THE DATA WERE COLLECTED FROM A MEROHEDRALLY TWINNED CRYSTAL | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: 1.75M AMMONIUM SULPHATE, 5MM 2-OXOGLUTARATE,0.1M HEPES PH7.5, pH 7.50 | ||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown / Details: Valegard, K., (1998) Nature, 394, 805. / PH range low: 7.5 / PH range high: 7 | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II  / Beamline: I711 / Wavelength: 1.092 / Beamline: I711 / Wavelength: 1.092 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Apr 15, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.092 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→56.8 Å / Num. obs: 30873 / % possible obs: 92.6 % / Redundancy: 4.5 % / Rmerge(I) obs: 0.085 |
| Reflection shell | Resolution: 1.7→1.76 Å / Rmerge(I) obs: 0.231 / % possible all: 99 |
| Reflection | *PLUS Highest resolution: 1.7 Å / Num. measured all: 216411 / Rmerge(I) obs: 0.087 |
| Reflection shell | *PLUS % possible obs: 99 % / Rmerge(I) obs: 0.231 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1RXF Resolution: 1.7→56.8 Å / SU B: 2.404 / SU ML: 0.081 / Cross valid method: THROUGHOUT / ESU R: 0.122 / ESU R Free: 0.125 Details: THE RESIDUES MISSING IN THE PDB ENTRY (80-96,168-177 AND 310-311) ARE DISORDERED, AND THEREFORE OMITTED FROM THE MODEL
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 32.202 Å2
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→56.8 Å
| ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.7 Å / Rfactor Rfree: 0.24 / Rfactor Rwork: 0.188 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS |
