 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1n3y | ||||||
|---|---|---|---|---|---|---|---|
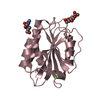
| Title | Crystal structure of the alpha-X beta2 integrin I domain | ||||||
 Components Components | Integrin alpha-X | ||||||
 Keywords Keywords |  CELL ADHESION / alpha/beta Rossmann fold CELL ADHESION / alpha/beta Rossmann fold | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of endothelial tube morphogenesis / integrin alphaX-beta2 complex / positive regulation of myelination / heterotypic cell-cell adhesion / integrin complex / cell adhesion mediated by integrin / tertiary granule membrane / ficolin-1-rich granule membrane / ECM proteoglycans / Integrin cell surface interactions ...positive regulation of endothelial tube morphogenesis / integrin alphaX-beta2 complex / positive regulation of myelination / heterotypic cell-cell adhesion / integrin complex / cell adhesion mediated by integrin / tertiary granule membrane / ficolin-1-rich granule membrane / ECM proteoglycans / Integrin cell surface interactions / cell-matrix adhesion / secretory granule membrane / integrin-mediated signaling pathway / Cell surface interactions at the vascular wall / animal organ morphogenesis / receptor tyrosine kinase binding / cell-cell adhesion / positive regulation of angiogenesis / integrin binding / signaling receptor activity / Interleukin-4 and Interleukin-13 signaling / defense response to virus / cell adhesion / positive regulation of cell migration / external side of plasma membrane / Neutrophil degranulation / positive regulation of cell population proliferation / positive regulation of gene expression / cell surface / membrane / metal ion binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.65 Å | ||||||
 Authors Authors | Vorup-Jensen, T. / Ostermeier, C. / Shimaoka, M. / Hommel, U. / Springer, T.A. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2003 Title: Structure and allosteric regulation of the alpha X beta 2 integrin I domain. Authors: Vorup-Jensen, T. / Ostermeier, C. / Shimaoka, M. / Hommel, U. / Springer, T.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1n3y.cif.gz | 57 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1n3y.ent.gz | 41 KB | Display | PDB format |
| PDBx/mmJSON format | 1n3y.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n3/1n3yftp://data.pdbj.org/pub/pdb/validation_reports/n3/1n3y | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22355.283 Da / Num. of mol.: 1 / Fragment: I domain / Mutation: T192S, T234A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ITGAX OR CD11C / Plasmid: pET28a / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P20702 Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P20702 |
|---|---|
| #2: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 291 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 291 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 54.03 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5 Details: ammonium sulfate, glycerol, sodium acetate, EDTA, pH 5.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS / Wavelength: 1.54178 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 18, 2001 / Details: osmic mirrors |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54178 Å / Relative weight: 1 |
| Reflection | Resolution: 1.65→30 Å / Num. obs: 32357 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 5.4 % / Biso Wilson estimate: 20.7 Å2 / Rmerge(I) obs: 0.061 / Net I/σ(I): 14.9 |
| Reflection shell | Resolution: 1.65→1.71 Å / Rmerge(I) obs: 0.229 / Num. unique all: 3199 / % possible all: 100 |
| Reflection | *PLUS Lowest resolution: 30 Å / Num. obs: 32389 / Num. measured all: 278307 |
| Reflection shell | *PLUS % possible obs: 100 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: In-house structure of a Mac1 I domain Resolution: 1.65→27.72 Å / Rfactor Rfree error: 0.006 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 61.5352 Å2 / ksol: 0.412969 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.5 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.65→27.72 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.65→1.75 Å / Rfactor Rfree error: 0.018 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 2 % | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
