 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1m9s: Crystal structure of Internalin B (InlB), a Listeria monocytogene... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1m9s | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Internalin B (InlB), a Listeria monocytogenes virulence protein containing SH3-like domains. | ||||||
 Components Components | Internalin B | ||||||
 Keywords Keywords |  SIGNALING PROTEIN / internalin / cell invasion / GW domains / SH3 domains SIGNALING PROTEIN / internalin / cell invasion / GW domains / SH3 domains | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan-based cell wall / InlB-mediated entry of Listeria monocytogenes into host cell / heparin binding / lipid binding / cell surface / extracellular region / metal ion binding / plasma membrane / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Listeria monocytogenes (bacteria) Listeria monocytogenes (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.65 Å | ||||||
 Authors Authors | Marino, M. / Banerjee, M. / Jonquieres, R. / Cossart, P. / Ghosh, P. | ||||||
 Citation Citation | Journal: Embo J. / Year: 2002 Title: GW domains of the Listeria monocytogenes invasion protein InlB are SH3-like and mediate binding to host ligands Authors: Marino, M. / Banerjee, M. / Jonquieres, R. / Cossart, P. / Ghosh, P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1m9s.cif.gz | 120.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1m9s.ent.gz | 91.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1m9s.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/m9/1m9sftp://data.pdbj.org/pub/pdb/validation_reports/m9/1m9s | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |
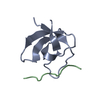







-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
| ||||||||
| Details | The biological assembly is a monomer composed of InlB residues 36 to 630. Residues 320 to 390 are present but disordered. Ambiguity in the position of this disordered region means that the monomer is made up of one N-terminal domain and one C-terminal domain, but there are two possible positions for the C-terminal domain. The alternate position for C-terminal domain is generated by the two fold axis (x = -x, y = y, z = 1/2 - z). |
-Components
| #1: Protein | Mass: 68622.086 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Listeria monocytogenes (bacteria) / Gene: inlB / Plasmid: pET28b / Species (production host): Escherichia coli / Production host: Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P25147, UniProt: P0DQD2*PLUS | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-TB /   Mass: 158.925 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Tb Mass: 158.925 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Tb#3: Chemical | ChemComp-SO4 / | Sulfate  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 65 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 65 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.33 Å3/Da / Density % sol: 76.93 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: lithium sulfate, glycerol, dithiothreitol, MES, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||||||||
| Detector |
| ||||||||||||||||||||||||
| Radiation | Monochromator: Si (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||||||||
| Reflection | Resolution: 2.65→50 Å / Num. all: 43356 / Num. obs: 37423 / % possible obs: 86.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Rmerge(I) obs: 0.081 / Net I/σ(I): 14.4 | ||||||||||||||||||||||||
| Reflection shell | Resolution: 2.65→2.74 Å / Rmerge(I) obs: 0.561 / Mean I/σ(I) obs: 1.53 / % possible all: 59 | ||||||||||||||||||||||||
| Reflection | *PLUS Lowest resolution: 50 Å / Redundancy: 4.2 % / Rmerge(I) obs: 0.081 | ||||||||||||||||||||||||
| Reflection shell | *PLUS % possible obs: 59 % / Rmerge(I) obs: 0.561 / Mean I/σ(I) obs: 1.5 |

- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.65→50 Å / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 93.46 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.65→50 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 50 Å / % reflection Rfree: 5 % / Rfactor obs: 0.274 / Rfactor Rfree: 0.302 / Rfactor Rwork: 0.273 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.65 Å / Lowest resolution: 2.74 Å / Rfactor Rfree: 0.488 / Rfactor Rwork: 0.46 |
