 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jvy: Maltodextrin-binding protein variant D207C/A301GS/P316C with beta... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jvy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Maltodextrin-binding protein variant D207C/A301GS/P316C with beta-mercaptoethanol mixed disulfides | |||||||||
 Components Components | maltodextrin-binding protein | |||||||||
 Keywords Keywords |  TRANSPORT PROTEIN / intermolecular / cross-link / disulfide TRANSPORT PROTEIN / intermolecular / cross-link / disulfide | |||||||||
| Function / homology |  Function and homology information Function and homology informationdetection of maltose stimulus / maltose binding / maltose transport complex / maltose transport / maltodextrin transmembrane transport / carbohydrate transmembrane transporter activity / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / carbohydrate transport / ATP-binding cassette (ABC) transporter complex / cell chemotaxis ...detection of maltose stimulus / maltose binding / maltose transport complex / maltose transport / maltodextrin transmembrane transport / carbohydrate transmembrane transporter activity / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / carbohydrate transport / ATP-binding cassette (ABC) transporter complex / cell chemotaxis / outer membrane-bounded periplasmic space / periplasmic space / DNA damage response / membraneSimilarity search - Function | |||||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | |||||||||
 Authors Authors | Srinivasan, U. / Iyer, G.H. / Przybycien, T.A. / Samsonoff, W.A. / Bell, J.A. | |||||||||
 Citation Citation | Journal: Protein Eng. / Year: 2002 Title: Crystine: Fibrous Biomolecular Material from Protein Crystals Cross-linked in a Specific Geometry Authors: Srinivasan, U. / Iyer, G.H. / Przybycien, T.A. / Samsonoff, W.A. / Bell, J.A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jvy.cif.gz | 96.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jvy.ent.gz | 71.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1jvy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jv/1jvyftp://data.pdbj.org/pub/pdb/validation_reports/jv/1jvy | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1jvxC  1mdqS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41103.719 Da / Num. of mol.: 1 / Mutation: D207C/A301GS/P316C Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Gene: malE / Plasmid: pMAL-c2 / Production host: Escherichia coli (E. coli) / Strain (production host): ER2058 / References: UniProt: P02928, UniProt: P0AEX9*PLUS |
|---|---|
| #2: Polysaccharide | alpha-D-glucopyranose-(1-4)-alpha-D-glucopyranose / alpha-maltose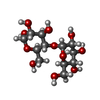  , Oligosaccharide / Class: Nutrient / Mass: 342.297 Da / Num. of mol.: 1 , Oligosaccharide / Class: Nutrient / Mass: 342.297 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: alpha-maltose |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 371 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 371 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.71 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 6.2 Details: PEG 6000, sodium MES, maltose, sodium azide, beta-mercaptoethanol, pH 6.2, VAPOR DIFFUSION, HANGING DROP, temperature 277K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 0.98 Å / Beamline: X12C / Wavelength: 0.98 Å |
| Detector | Type: BRANDEIS - B1 / Detector: CCD / Date: Jun 6, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→23.86 Å / Num. all: 28657 / Num. obs: 28657 / % possible obs: 100 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.4 % / Biso Wilson estimate: 16.9 Å2 / Rmerge(I) obs: 0.06 |
| Reflection shell | Resolution: 1.9→2.5 Å / Mean I/σ(I) obs: 5.6 / % possible all: 100 |
| Reflection | *PLUS Lowest resolution: 9999 Å / % possible obs: 100 % / Num. measured all: 127321 / Rmerge(I) obs: 0.06 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1MDQ Resolution: 1.9→23.86 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 1416969.77 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: BABINET / Bsol: 280 Å2 / ksol: 0.9 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 30.8 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→23.86 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→2.02 Å / Rfactor Rfree error: 0.014 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNX / Version: 2000.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 9.9 % / Rfactor obs: 0.207 | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 30.8 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.303 / % reflection Rfree: 10.3 % / Rfactor Rwork: 0.231 |
