 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jvn: CRYSTAL STRUCTURE OF IMIDAZOLE GLYCEROL PHOSPHATE SYNTHASE: A TUN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jvn | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF IMIDAZOLE GLYCEROL PHOSPHATE SYNTHASE: A TUNNEL THROUGH A (BETA/ALPHA)8 BARREL JOINS TWO ACTIVE SITES | ||||||
 Components Components | BIFUNCTIONAL HISTIDINE BIOSYNTHESIS PROTEIN HISHF | ||||||
 Keywords Keywords |  TRANSFERASE / substrate channeling / amidotransferase / TIM-barrel as a substrate tunnel TRANSFERASE / substrate channeling / amidotransferase / TIM-barrel as a substrate tunnel | ||||||
| Function / homology |  Function and homology information Function and homology informationimidazole glycerol-phosphate synthase / imidazoleglycerol-phosphate synthase activity / oxo-acid-lyase activity / glutaminase / L-histidine biosynthetic process / glutaminase activity / glutamine metabolic process / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.1 Å | ||||||
 Authors Authors | Chaudhuri, B.N. / Smith, J.L. / Davisson, V.J. / Myers, R.S. / Lange, S.C. / Chittur, S.V. | ||||||
 Citation Citation | Journal: Structure / Year: 2001 Title: Crystal structure of imidazole glycerol phosphate synthase: a tunnel through a (beta/alpha)8 barrel joins two active sites. Authors: Chaudhuri, B.N. / Lange, S.C. / Myers, R.S. / Chittur, S.V. / Davisson, V.J. / Smith, J.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jvn.cif.gz | 219.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jvn.ent.gz | 183.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1jvn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jv/1jvnftp://data.pdbj.org/pub/pdb/validation_reports/jv/1jvn | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| 2 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Details | Monomer in solution |
-Components
| #1: Protein | Mass: 61564.273 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: PROTEIN RESIDUE 143, NUMBER 83, IS CYS COVALENTLY MODIFIED BY ACIVICIN Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Gene: HIS7 / Plasmid: PHIS7-T7 / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / Variant (production host): BL21(DE3) Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / Variant (production host): BL21(DE3)References: UniProt: P33734, Transferases; Glycosyltransferases; Pentosyltransferases#2: Chemical | Nickel  Mass: 58.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ni Mass: 58.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ni#3: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-POP / | Pyrophosphate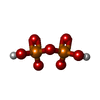  Mass: 175.959 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H2O7P2 Mass: 175.959 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H2O7P2#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 412 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 412 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.61 Å3/Da / Density % sol: 52.93 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 28 % PEG MME 5000, 0.2 M Ammonium sulfate, 0.1 M MES buffer, pH 6.5, VAPOR DIFFUSION, HANGING DROP at 298K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / PH range low: 7 / PH range high: 6.5 | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| |||||||||||||||
| Detector |
| |||||||||||||||
| Radiation |
| |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 2.1→100 Å / Num. obs: 73741 / % possible obs: 100 % / Observed criterion σ(I): 2 / Redundancy: 14.3 % / Biso Wilson estimate: 21.3 Å2 / Rmerge(I) obs: 0.067 / Net I/σ(I): 7.3 | |||||||||||||||
| Reflection shell | Resolution: 2.1→2.18 Å / Redundancy: 13.1 % / Rmerge(I) obs: 0.39 / Mean I/σ(I) obs: 2 / % possible all: 99.9 | |||||||||||||||
| Reflection | *PLUS Lowest resolution: 100 Å / % possible obs: 100 % | |||||||||||||||
| Reflection shell | *PLUS % possible obs: 99.9 % |

- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.1→75.21 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 1768716.76 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: CNS dictionary
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 32.9268 Å2 / ksol: 0.363298 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.1 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→75.21 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.23 Å / Rfactor Rfree error: 0.014 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 31.1 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.305 / % reflection Rfree: 4.9 % / Rfactor Rwork: 0.28 |
