 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hvv: SELF-ASSOCIATION OF THE H3 REGION OF SYNTAXIN 1A: IMPLICATIONS FO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hvv | ||||||
|---|---|---|---|---|---|---|---|
| Title | SELF-ASSOCIATION OF THE H3 REGION OF SYNTAXIN 1A: IMPLICATIONS FOR SNARE COMPLEX ASSEMBLY | ||||||
 Components Components | SYNTAXIN 1A STX1A STX1A | ||||||
 Keywords Keywords | ENDOCYTOSIS/EXOCYTOSIS / four-helix bundle / homotetramer / alpha-helix / ENDOCYTOSIS-EXOCYTOSIS COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationmyosin head/neck binding / Other interleukin signaling / synaptic vesicle fusion to presynaptic active zone membrane / synaptobrevin 2-SNAP-25-syntaxin-1a-complexin II complex / synaptobrevin 2-SNAP-25-syntaxin-1a complex / synaptobrevin 2-SNAP-25-syntaxin-1a-complexin I complex / positive regulation of norepinephrine secretion / regulation of synaptic vesicle priming / Glutamate Neurotransmitter Release Cycle / Norepinephrine Neurotransmitter Release Cycle ...myosin head/neck binding / Other interleukin signaling / synaptic vesicle fusion to presynaptic active zone membrane / synaptobrevin 2-SNAP-25-syntaxin-1a-complexin II complex / synaptobrevin 2-SNAP-25-syntaxin-1a complex / synaptobrevin 2-SNAP-25-syntaxin-1a-complexin I complex / positive regulation of norepinephrine secretion / regulation of synaptic vesicle priming / Glutamate Neurotransmitter Release Cycle / Norepinephrine Neurotransmitter Release Cycle / Acetylcholine Neurotransmitter Release Cycle / positive regulation of catecholamine secretion / Serotonin Neurotransmitter Release Cycle / GABA synthesis, release, reuptake and degradation / regulated exocytosis / Dopamine Neurotransmitter Release Cycle / synaptic vesicle docking / response to gravity / Insertion of tail-anchored proteins into the endoplasmic reticulum membrane / positive regulation of calcium ion-dependent exocytosis / vesicle docking / chloride channel inhibitor activity / SNARE complex / SNAP receptor activity / secretion by cell / LGI-ADAM interactions / hormone secretion / calcium-ion regulated exocytosis / actomyosin / regulation of exocytosis / ATP-dependent protein binding / neurotransmitter transport / protein localization to membrane / SNARE complex assembly / positive regulation of neurotransmitter secretion / insulin secretion / myosin binding / modulation of excitatory postsynaptic potential / exocytosis / positive regulation of exocytosis / synaptic vesicle exocytosis / positive regulation of excitatory postsynaptic potential / protein sumoylation / synaptic vesicle endocytosis / endomembrane system / calcium channel inhibitor activity / presynaptic active zone membrane / SNARE binding / acrosomal vesicle / secretory granule / postsynaptic density membrane / intracellular protein transport / Schaffer collateral - CA1 synapse / synaptic vesicle membrane / kinase binding / calcium-dependent protein binding / protein-macromolecule adaptor activity / synaptic vesicle / presynapse / presynaptic membrane / nuclear membrane / transmembrane transporter binding / postsynaptic density / neuron projection / axon / synapse / glutamatergic synapse / protein-containing complex binding / protein-containing complex / membrane / identical protein binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Rattus norvegicus (Norway rat) Rattus norvegicus (Norway rat) | ||||||
| Method | X-RAY DIFFRACTION / MAD / Resolution: 2.4 Å | ||||||
 Authors Authors | Misura, K.M.S. / Scheller, R.H. / Weis, W.I. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2001 Title: Self-association of the H3 region of syntaxin 1A. Implications for intermediates in SNARE complex assembly. Authors: Misura, K.M. / Scheller, R.H. / Weis, W.I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hvv.cif.gz | 62.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hvv.ent.gz | 47.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1hvv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hv/1hvvftp://data.pdbj.org/pub/pdb/validation_reports/hv/1hvv | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is an anti-parallel homotetramer contained within one asymmetric unit. |
-Components
| #1: Protein | STX1A / NEURON-SPECIFIC ANTIGEN HPC-1 / SYNAPTOTAGMIN ASSOCIATED 35 KDA PROTEIN Mass: 8857.096 Da / Num. of mol.: 4 / Fragment: RESIDUES 190-264 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Rattus norvegicus (Norway rat) / Gene: STX1A / Plasmid: PGEX-KG / Production host:  Escherichia coli (E. coli) / Strain (production host): AB1899 / References: UniProt: P32851 Escherichia coli (E. coli) / Strain (production host): AB1899 / References: UniProt: P32851#2: Chemical | ChemComp-TAR / | Tartaric acid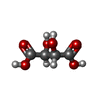  Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.67 Å3/Da / Density % sol: 65 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5 Details: sodium potassium tartrate, sodium acetate, pH 5.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 25 ℃ / pH: 8.4 | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.54 Å |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Apr 3, 1995 / Details: mirrors |
| Radiation | Monochromator: Graphite / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→45 Å / Num. all: 21053 / Num. obs: 20870 / % possible obs: 99.2 % / Observed criterion σ(F): 3 / Observed criterion σ(I): 3 / Redundancy: 3.8 % / Biso Wilson estimate: 28 Å2 / Rmerge(I) obs: 0.069 / Net I/σ(I): 19.8 |
| Reflection shell | Resolution: 2.4→45 Å / Redundancy: 3.2 % / Rmerge(I) obs: 0.313 / % possible all: 99.1 |
| Reflection | *PLUS Redundancy: 4.4 % / Rmerge(I) obs: 0.054 |
| Reflection shell | *PLUS % possible obs: 99.1 % / Redundancy: 3.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.4→30 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 2243567.95 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 36.2 Å2 / ksol: 0.351 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.7 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→30 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.47 Å / Rfactor Rfree error: 0.018 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 7.8 % | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 41.7 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.286 / % reflection Rfree: 7.8 % / Rfactor Rwork: 0.244 |
