 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ghh | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | SOLUTION STRUCTURE OF DINI | |||||||||
 Components Components | DNA-DAMAGE-INDUCIBLE PROTEIN I | |||||||||
 Keywords Keywords |  PROTEIN BINDING / bicelle / DinI / dipolar coupling / liquid crystal / Pf1 / RecA PROTEIN BINDING / bicelle / DinI / dipolar coupling / liquid crystal / Pf1 / RecA | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | |||||||||
| Method | SOLUTION NMR / simulated annealing, molecular dynamics in Cartesian space | |||||||||
 Authors Authors | Ramirez, B.E. / Voloshin, O.N. / Camerini-Otero, R.D. / Bax, A. | |||||||||
 Citation Citation | Journal: Protein Sci. / Year: 2000 Title: Solution structure of DinI provides insight into its mode of RecA inactivation. Authors: Ramirez, B.E. / Voloshin, O.N. / Camerini-Otero, R.D. / Bax, A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
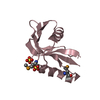
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ghh.cif.gz | 482.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ghh.ent.gz | 404.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1ghh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gh/1ghhftp://data.pdbj.org/pub/pdb/validation_reports/gh/1ghh | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: Protein | Mass: 8957.937 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Plasmid: PET21A / Production host: Escherichia coli (E. coli) / References: UniProt: P0ABR1 |
|---|
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NMR experiment |
| ||||||||||||||||||||||||||||||||||||
| NMR details | Text: This structure was determined using two sets of dipolar coupling restraints. One set was recorded in a bicelle liquid crystal solution; the other set was recorded in a phage liquid crystal ...Text: This structure was determined using two sets of dipolar coupling restraints. One set was recorded in a bicelle liquid crystal solution; the other set was recorded in a phage liquid crystal solution. See citation 1 for details on solution conditions. A total of 127 N-H, 135 CA-HA, 138 CA-CO, 61 N-CO, and 64 CO-HN dipolar restraints were used in the structure calculation. Additional restraints included 592 intraresidue, 278 short range, 104 medium range, and 140 long range NOE restraints as well as 76 phi, 51 psi, and 21 chi1 dihedral restraints. A conformational database was employed in the simulated annealing calculation. No radius of gyration term was employed in the simulated annealing calculation. |
- Sample preparation
Sample preparation
| Details |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample conditions |
| ||||||||||||||||
| Crystal grow | *PLUS Method: other / Details: NMR |
-NMR measurement
| NMR spectrometer |
|
|---|
- Processing
Processing
| NMR software |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method: simulated annealing, molecular dynamics in Cartesian space Software ordinal: 1 Details: Structures were calculated in a three stage process. In the first stage, folds were calculated from a fully-extended chain based only on torsion and NOE restraints. In the second stage, the ...Details: Structures were calculated in a three stage process. In the first stage, folds were calculated from a fully-extended chain based only on torsion and NOE restraints. In the second stage, the ten best structures of stage 1 were used as starting structures in a simulated annealing calculation that included dipolar restraints measured in bicelles. In the last stage, the ten lowest energy structures of Stage 2 were used as starting structures in a simulated annealing calculation that also included dipolar restraints measured in a phage liquid crystal. | |||||||||||||||
| NMR ensemble | Conformer selection criteria: structures with the lowest energy Conformers calculated total number: 30 / Conformers submitted total number: 20 |
