 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1g6u | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A DOMAIN SWAPPED DIMER | ||||||
 Components Components | DOMAIN SWAPPED DIMER | ||||||
 Keywords Keywords |  DE NOVO PROTEIN / designed three helix bundle DE NOVO PROTEIN / designed three helix bundle | ||||||
| Function / homology | Helix hairpin bin / Helix Hairpins / Orthogonal Bundle / Mainly Alpha / trifluoroacetic acid Function and homology information Function and homology information | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 1.48 Å | ||||||
 Authors Authors | Ogihara, N.L. / Ghirlanda, G. / Bryson, J.W. / Gingery, M. / DeGrado, W.F. / Eisenberg, D. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2001 Title: Design of three-dimensional domain-swapped dimers and fibrous oligomers. Authors: Ogihara, N.L. / Ghirlanda, G. / Bryson, J.W. / Gingery, M. / DeGrado, W.F. / Eisenberg, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1g6u.cif.gz | 28.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1g6u.ent.gz | 21 KB | Display | PDB format |
| PDBx/mmJSON format | 1g6u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g6/1g6uftp://data.pdbj.org/pub/pdb/validation_reports/g6/1g6u | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|







-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| 2 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein/peptide | Mass: 5131.009 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: The peptide was chemically synthesized. #2: Chemical | ChemComp-SO4 / | Sulfate  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-TFA / | Trifluoroacetic acid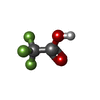  Mass: 114.023 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2HF3O2 Mass: 114.023 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2HF3O2#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.13 Å3/Da / Density % sol: 42.32 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion / pH: 6.5 Details: 100 mM MES, pH 6.5, 100 mM NaCl, 2.6 M ammonium sulfate, 1% dioxane, VAPOR DIFFUSION, temperature 298K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12B / Wavelength: 0.975 Å / Beamline: X12B / Wavelength: 0.975 Å |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Details: mirrors |
| Radiation | Monochromator: mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.975 Å / Relative weight: 1 |
| Reflection | Resolution: 1.48→30 Å / Num. all: 14554 / Num. obs: 14554 / % possible obs: 97.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 5.5 % / Biso Wilson estimate: 17.5 Å2 / Rmerge(I) obs: 0.082 |
| Reflection shell | Resolution: 1.48→1.57 Å / Rmerge(I) obs: 0.289 / Num. unique all: 2129 / % possible all: 88 |
| Reflection | *PLUS Num. measured all: 80427 |
| Reflection shell | *PLUS % possible obs: 94.1 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 1.48→30 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.5 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.48→30 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.48→1.57 Å / Rfactor Rfree error: 0.022 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR(ONLINE) / Version: 3.82 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 10.1 % | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 16.5 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.314 / % reflection Rfree: 9.2 % / Rfactor Rwork: 0.285 |
