 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1f6m: CRYSTAL STRUCTURE OF A COMPLEX BETWEEN THIOREDOXIN REDUCTASE, THI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1f6m | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A COMPLEX BETWEEN THIOREDOXIN REDUCTASE, THIOREDOXIN, AND THE NADP+ ANALOG, AADP+ | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  OXIDOREDUCTASE / ALTERNATE CONFORMATION / TERNARY COMPLEX / DOMAIN MOTION / redox-active center / NADP / FAD / electron transport OXIDOREDUCTASE / ALTERNATE CONFORMATION / TERNARY COMPLEX / DOMAIN MOTION / redox-active center / NADP / FAD / electron transport | ||||||
| Function / homology |  Function and homology informationthioredoxin-disulfide reductase complex / thioredoxin-disulfide reductase / thioredoxin-disulfide reductase (NADPH) activity / DNA polymerase processivity factor activity / protein-disulfide reductase activity / removal of superoxide radicals / cell redox homeostasis / flavin adenine dinucleotide binding / cytosol / cytoplasm Function and homology informationthioredoxin-disulfide reductase complex / thioredoxin-disulfide reductase / thioredoxin-disulfide reductase (NADPH) activity / DNA polymerase processivity factor activity / protein-disulfide reductase activity / removal of superoxide radicals / cell redox homeostasis / flavin adenine dinucleotide binding / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.95 Å | ||||||
 Authors Authors | Lennon, B.W. / Williams Jr., C.H. / Ludwig, M.L. | ||||||
 Citation Citation | Journal: Science / Year: 2000 Title: Twists in catalysis: alternating conformations of Escherichia coli thioredoxin reductase. Authors: Lennon, B.W. / Williams Jr., C.H. / Ludwig, M.L. #1: Journal: J.Mol.Biol. / Year: 1994Title: Crystal structure of Escherichia coli thioredoxin reductase refined at 2 A resolution. Implications for a large conformational change during catalysis. Authors: Waksman, G. / Krishna, T.S. / Sweet, R.M. / Williams Jr., C.H. / Kuriyan, J. #2: Journal: Nature / Year: 1991Title: Convergent evolution of similar function in two structurally divergent enzymes Authors: Kuriyan, J. / Krishna, T.S. / Wong, L. / Guenther, B. / Pahler, A. / Williams Jr., C.H. / Model, P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1f6m.cif.gz | 344 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1f6m.ent.gz | 280.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1f6m.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f6/1f6mftp://data.pdbj.org/pub/pdb/validation_reports/f6/1f6m | HTTPS FTP |
|---|
-Related structure data

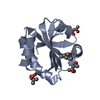











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| 2 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
| |||||||||||||||
| Details | The biological assembly of thioreodxin reductase is a dimer consisting of chains A and B (corresponding to thioredoxin reductase chains B and A in the primary citation). This structure includes one FAD cofactor and one pyridine nucleotide product analog (AADP+) molecule per enzyme chain. The corresponding covalently bound thioredoxin substrate molecules (one per enzyme monomer) are chains C and D (corresponding to thioredoxin chains B and A in the primary citation). / The biological assembly of thioredoxin reductase is a dimer consisting of chains E and F (corresponding to thioredoxin reductase chains D and C in the primary citation). This structure includes one FAD cofactor and one pyridine nucleotide product analog (AADP+) molecule per enzyme chain. The corresponding covalently bound thioredoxin substrate molecules (one per enzyme monomer) are chains G and H (corresponding to thioredoxin chains D and C in the primary citation). |
-Components
| #1: Protein | Mass: 34513.703 Da / Num. of mol.: 4 / Mutation: C135S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Production host: Escherichia coli (E. coli) / References: UniProt: P0A9P4, EC: 1.6.4.5#2: Protein | Mass: 11671.322 Da / Num. of mol.: 4 / Mutation: C35S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Production host: Escherichia coli (E. coli) / References: UniProt: P0AA25#3: Chemical | ChemComp-FAD / Flavin adenine dinucleotide  Mass: 785.550 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H33N9O15P2 Mass: 785.550 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H33N9O15P2Details: Purchased from Sigma Chemical Company, St. Louis, MO Comment: FAD*YM #4: Chemical | ChemComp-3AA / 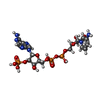  Mass: 716.403 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C20H29N7O16P3 Mass: 716.403 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C20H29N7O16P3#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.91 Å3/Da / Density % sol: 58.45 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 6 Details: cacodylate, ammonium sulfate, PEG 3350, 3-aminopyridine adenine dinucleotide phosphate, pH 6.0, VAPOR DIFFUSION, HANGING DROP, temperature 295K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / pH: 7 | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 113 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Sep 16, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.95→31.38 Å / Num. all: 598907 / Num. obs: 45732 / % possible obs: 99.8 % / Observed criterion σ(I): 3.4 / Redundancy: 13.1 % / Biso Wilson estimate: 140.2 Å2 / Rmerge(I) obs: 0.084 / Net I/σ(I): 15 |
| Reflection shell | Resolution: 2.95→3.06 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.358 / Mean I/σ(I) obs: 3.4 / Num. unique all: 4492 / % possible all: 99 |
| Reflection | *PLUS Num. measured all: 598907 |
| Reflection shell | *PLUS % possible obs: 99 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1TRB, 2TRX Resolution: 2.95→31.38 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 2194524.24 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: several solvent-exposed regions of the thioredoxin chains cannot be modeled from the density. These regions are apparent from B factors >100 A2 or atom occupancies of 0.5. They include ...Details: several solvent-exposed regions of the thioredoxin chains cannot be modeled from the density. These regions are apparent from B factors >100 A2 or atom occupancies of 0.5. They include residues 1-20 in chains C,G; 1-22 in chains D,H; residues 61-62 and 81-85 in chains D,H and other side chains.
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 25.11 Å2 / ksol: 0.304 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.2 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.95→31.38 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Refine-ID: X-RAY DIFFRACTION / Weight Biso : 2 / Weight position: 150
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.95→3.06 Å / Rfactor Rfree error: 0.022 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 6 % / Rfactor obs: 0.205 | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 46.2 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.369 / % reflection Rfree: 6.1 % / Rfactor Rwork: 0.323 |
