 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dhf: CRYSTAL STRUCTURES OF RECOMBINANT HUMAN DIHYDROFOLATE REDUCTASE C... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dhf | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURES OF RECOMBINANT HUMAN DIHYDROFOLATE REDUCTASE COMPLEXED WITH FOLATE AND 5-DEAZOFOLATE | ||||||
 Components Components | DIHYDROFOLATE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / OXIDO-REDUCTASE | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of removal of superoxide radicals / tetrahydrobiopterin biosynthetic process / Metabolism of folate and pterines / tetrahydrofolate metabolic process / sequence-specific mRNA binding / response to methotrexate / axon regeneration / folic acid binding / dihydrofolate metabolic process / G1/S-Specific Transcription ...regulation of removal of superoxide radicals / tetrahydrobiopterin biosynthetic process / Metabolism of folate and pterines / tetrahydrofolate metabolic process / sequence-specific mRNA binding / response to methotrexate / axon regeneration / folic acid binding / dihydrofolate metabolic process / G1/S-Specific Transcription / glycine biosynthetic process / dihydrofolate reductase / folic acid metabolic process / dihydrofolate reductase activity / NADPH binding / Tetrahydrobiopterin (BH4) synthesis, recycling, salvage and regulation / tetrahydrofolate biosynthetic process / mRNA regulatory element binding translation repressor activity / positive regulation of nitric-oxide synthase activity / one-carbon metabolic process / NADP binding / negative regulation of translation / mRNA binding / mitochondrion / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 2.3 Å | ||||||
 Authors Authors | Davies /II, J.F. / Kraut, J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1990 Title: Crystal structures of recombinant human dihydrofolate reductase complexed with folate and 5-deazafolate. Authors: Davies 2nd., J.F. / Delcamp, T.J. / Prendergast, N.J. / Ashford, V.A. / Freisheim, J.H. / Kraut, J. #1: Journal: Biochemistry / Year: 1988Title: Expression and Site-Directed Mutagenesis of Human Dihydrofolate Reductase Authors: Prendergast, N.J. / Delcamp, T.J. / Smith, P.L. / Freisheim, J.H. | ||||||
| History |
| ||||||
| Remark 650 | HELIX IN EACH CHAIN, RESIDUES ASP 21 - LEU 22 - PRO 23 - TRP 24 - PRO 25 - PRO 26 ARE IN LEFT- ...HELIX IN EACH CHAIN, RESIDUES ASP 21 - LEU 22 - PRO 23 - TRP 24 - PRO 25 - PRO 26 ARE IN LEFT-HANDED POLYPROLINE HELIX CONFORMATION. | ||||||
| Remark 700 | SHEET THE FOLLOWING REMARKS APPLY TO EACH CHAIN. IN THE *HELIX*, *SHEET* AND *TURN* RECORDS BELOW, ...SHEET THE FOLLOWING REMARKS APPLY TO EACH CHAIN. IN THE *HELIX*, *SHEET* AND *TURN* RECORDS BELOW, AN *A* OR *B* HAS BEEN APPENDED TO THE NAMES USED IN THIS REMARK TO DISTINGUISH CHAINS. RESIDUE GLN 102 PARTICIPATES IN BOTH HELIX E AND EP. RESIDUES LYS 108 AND VAL 109 PARTICIPATE IN BOTH HELIX EP AND STRAND E. RESIDUE GLU 172 IS IN TIGHT-TURN 8 AND BETA STRAND G. RESIDUE ILE 175 IS IN TIGHT-TURN 8 AND BETA STRAND H. RESIDUES ASP 110 - MET 111 FORM A BETA-BULGE IN STRAND E. RESIDUES VAL 115 - GLY 116 FORM A BETA-BULGE IN STRAND E. TIGHT TURN 7 DISRUPTS STRAND G. THIS IS REPRESENTED ON THE SHEET RECORDS BELOW BY PRESENTING THE SHEET TWICE WITH STRAND 8 DIFFERENT. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dhf.cif.gz | 88.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dhf.ent.gz | 68.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1dhf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dh/1dhfftp://data.pdbj.org/pub/pdb/validation_reports/dh/1dhf | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUES PRO A 66 AND PRO B 66 ARE CIS PROLINES. 2: IN EACH CHAIN, THE PEPTIDE BOND LINKING GLY 116 TO GLY 117 IS IN THE CIS CONFORMATION. |
-Components
| #1: Protein | Mass: 21349.525 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / References: UniProt: P00374, dihydrofolate reductase#2: Chemical | Folate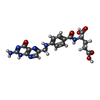  Mass: 441.397 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H19N7O6 Mass: 441.397 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H19N7O6#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47.36 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 5.9 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.3 Å / Num. obs: 16023 / % possible obs: 91 % / Rmerge(I) obs: 0.056 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor obs: 0.176 / Highest resolution: 2.3 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 2.3 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.3 Å / Rfactor obs: 0.176 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
