 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cko: STRUCTURE OF MRNA CAPPING ENZYME IN COMPLEX WITH THE CAP ANALOG GPPPG -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cko | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF MRNA CAPPING ENZYME IN COMPLEX WITH THE CAP ANALOG GPPPG | ||||||
 Components Components | MRNA CAPPING ENZYME | ||||||
 Keywords Keywords | CAPPING ENZYME / MRNA / NUCLEOTIDYLTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology information7-methylguanosine mRNA capping / mRNA guanylyltransferase activity / mRNA guanylyltransferase / GTP binding / ATP binding Similarity search - Function | ||||||
| Biological species |   Paramecium bursaria Chlorella virus 1 Paramecium bursaria Chlorella virus 1 | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT. THE ROTATION FUNCTION WAS SOLVED FOR EACH OF THE TWO DOMAINS SEPARATELY. TRANSLATION WAS PERFORMED WITH THE TWO DOMAINS INDEPENDENTLY BUT SIMULTANEOUSLY. / Resolution: 3.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT. THE ROTATION FUNCTION WAS SOLVED FOR EACH OF THE TWO DOMAINS SEPARATELY. TRANSLATION WAS PERFORMED WITH THE TWO DOMAINS INDEPENDENTLY BUT SIMULTANEOUSLY. / Resolution: 3.1 Å | ||||||
 Authors Authors | Hakansson, K. / Wigley, D.B. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1998 Title: Structure of a complex between a cap analogue and mRNA guanylyl transferase demonstrates the structural chemistry of RNA capping. Authors: Hakansson, K. / Wigley, D.B. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1997Title: Crystallization of the RNA Guanylyltransferase of Chlorella Virus Pbcv-1 Authors: Doherty, A.J. / Hakansson, K. / Ho, C.K. / Shuman, S. / Wigley, D.B. #2: Journal: Cell(Cambridge,Mass.) / Year: 1997Title: X-Ray Crystallography Reveals a Large Conformational Change During Guanyl Transfer by Mrna Capping Enzymes Authors: Hakansson, K. / Doherty, A.J. / Shuman, S. / Wigley, D.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cko.cif.gz | 78.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cko.ent.gz | 59.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1cko.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1cko_validation.pdf.gz | 741.7 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1cko_full_validation.pdf.gz | 755.7 KB | Display | |
| Data in XML | 1cko_validation.xml.gz | 15.5 KB | Display | |
| Data in CIF | 1cko_validation.cif.gz | 20.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ck/1ckoftp://data.pdbj.org/pub/pdb/validation_reports/ck/1cko | HTTPS FTP |
-Related structure data
| Related structure data |  1ckmS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37884.992 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Paramecium bursaria Chlorella virus 1 / Genus: Chlorovirus / Production host:  |
|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
| #3: Chemical | ChemComp-GP3 / 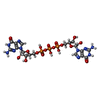  Mass: 788.406 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H27N10O18P3 Mass: 788.406 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H27N10O18P3 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.4 Å3/Da / Density % sol: 72 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 6.5 Details: HANGING DROP VAPOR DIFFUSION. 15 MG/ML PROTEIN IN 50 MM TRIS-HCL, 1.3 MM CAP ANALOG (GPPPG) 0.4 M NACL, 2 MM EDTA, 4 MM DTT, PH 7.5 WERE MIXED WITH AN EQUAL VOLUME OF AND EQUILIBRATED ...Details: HANGING DROP VAPOR DIFFUSION. 15 MG/ML PROTEIN IN 50 MM TRIS-HCL, 1.3 MM CAP ANALOG (GPPPG) 0.4 M NACL, 2 MM EDTA, 4 MM DTT, PH 7.5 WERE MIXED WITH AN EQUAL VOLUME OF AND EQUILIBRATED AGAINST 50 MM POTASSIUM PHOSPHATE, 5-10% PEG 8000, 2MM ZNCL2 PH 6.5., vapor diffusion - hanging drop PH range: 6.5-7.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX7.2 / Wavelength: 1.448 / Beamline: PX7.2 / Wavelength: 1.448 |
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Jun 1, 1997 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.448 Å / Relative weight: 1 |
| Reflection | Resolution: 3.1→20 Å / Num. obs: 12014 / % possible obs: 96.8 % / Observed criterion σ(I): 0 / Redundancy: 2.9 % / Biso Wilson estimate: 107 Å2 / Rmerge(I) obs: 0.035 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT. THE ROTATION FUNCTION WAS SOLVED FOR EACH OF THE TWO DOMAINS SEPARATELY. TRANSLATION WAS PERFORMED WITH THE TWO DOMAINS INDEPENDENTLY BUT SIMULTANEOUSLY. Starting model: OPEN FORM OF PDB ENTRY 1CKM Resolution: 3.1→10 Å / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 72 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.1→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
