 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1br1: SMOOTH MUSCLE MYOSIN MOTOR DOMAIN-ESSENTIAL LIGHT CHAIN COMPLEX W... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1br1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | SMOOTH MUSCLE MYOSIN MOTOR DOMAIN-ESSENTIAL LIGHT CHAIN COMPLEX WITH MGADP.ALF4 BOUND AT THE ACTIVE SITE | ||||||
 Components Components | (MYOSIN ) x 2 ) x 2 | ||||||
 Keywords Keywords | MUSCLE PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationmyosin II filament / myofibril assembly / elastic fiber assembly / myosin light chain binding / skeletal muscle myosin thick filament assembly / muscle myosin complex / myosin II binding / actomyosin / myosin filament / actomyosin structure organization ...myosin II filament / myofibril assembly / elastic fiber assembly / myosin light chain binding / skeletal muscle myosin thick filament assembly / muscle myosin complex / myosin II binding / actomyosin / myosin filament / actomyosin structure organization / myosin II complex / cardiac muscle cell development / structural constituent of muscle / microfilament motor activity / myofibril / myosin heavy chain binding / smooth muscle contraction / ADP binding / actin filament binding / actin binding / calmodulin binding / calcium ion binding / magnesium ion binding / ATP binding / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Gallus gallus (chicken) Gallus gallus (chicken) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.5 Å | ||||||
 Authors Authors | Dominguez, R. / Trybus, K.M. / Cohen, C. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 1998 Title: Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Authors: Dominguez, R. / Freyzon, Y. / Trybus, K.M. / Cohen, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1br1.cif.gz | 679.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1br1.ent.gz | 550.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1br1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/br/1br1ftp://data.pdbj.org/pub/pdb/validation_reports/br/1br1 | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj






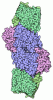




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||
| 4 | 
| ||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
-Protein , 2 types, 8 molecules ACEGBDFH
| #1: Protein | / MDE Mass: 93697.180 Da / Num. of mol.: 4 Fragment: CHAINS A, C, E, G, MOTOR DOMAIN, CHAINS B, D, F, H, ESSENTIAL LIGHT Source method: isolated from a genetically manipulated source Details: MG, ADP, ALF(4) / Source: (gene. exp.) Gallus gallus (chicken) / Tissue: SMOOTH MUSCLE / Cell line: SF9 / Organ: GIZZARD / Cell line (production host): SF9 / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P10587, EC: 3.6.1.32 Spodoptera frugiperda (fall armyworm) / References: UniProt: P10587, EC: 3.6.1.32#2: Protein | / MDEMass: 16873.025 Da / Num. of mol.: 4 Fragment: CHAINS A, C, E, G, MOTOR DOMAIN, CHAINS B, D, F, H, ESSENTIAL LIGHT Source method: isolated from a genetically manipulated source Details: MG, ADP, ALF(4) / Source: (gene. exp.) Gallus gallus (chicken) / Tissue: SMOOTH MUSCLE / Cell line: SF9 / Organ: GIZZARD / Cell line (production host): SF9 / Production host: Spodoptera frugiperda (fall armyworm) / References: UniProt: P02607, EC: 3.6.1.32 |
|---|
-Non-polymers , 4 types, 20 molecules 
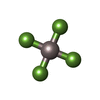


| #3: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#4: Chemical | ChemComp-ALF /  Mass: 102.975 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: AlF4 Mass: 102.975 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: AlF4#5: Chemical | ChemComp-ADP / Adenosine diphosphate Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4 Å3/Da / Density % sol: 70 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / pH: 7.2 Details: 1.8M AMMONIUM SULFATE 100MM HEPES PH 7.2 2MM MGADP 2MM AL(NO3)3 8MM NAF PROTEIN CONCENTRATION: 10MG/ML TEMPERATURE: 4 DEGREES CENTIGRADE, temperature 277K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.905 / Beamline: A1 / Wavelength: 0.905 |
| Detector | Detector: CCD / Date: May 1, 1997 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.905 Å / Relative weight: 1 |
| Reflection | Resolution: 3.5→20 Å / Num. obs: 79993 / % possible obs: 91.7 % / Redundancy: 4.7 % / Rsym value: 0.088 / Net I/σ(I): 21.6 |
| Reflection shell | Resolution: 3.5→3.82 Å / Redundancy: 3.3 % / Mean I/σ(I) obs: 9.8 / Rsym value: 0.301 / % possible all: 90.2 |
| Reflection | *PLUS Num. measured all: 375967 / Rmerge(I) obs: 0.088 |
| Reflection shell | *PLUS % possible obs: 90.2 % / Rmerge(I) obs: 0.301 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: SMOOTH MUSCLE MYOSIN MOTOR DOMAIN Resolution: 3.5→10 Å / Data cutoff high absF: 0 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 50.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error free: 0.6 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.5→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: RESTRAINTS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
