 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1b87 | ||||||
|---|---|---|---|---|---|---|---|
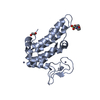
| タイトル | CRYSTAL STRUCTURE OF AN AMINOGLYCOSIDE 6'-N-ACETYLTRANSFERASE | ||||||
 要素 要素 | PROTEIN (AMINOGLYCOSIDE N6'-ACETYLTRANSFERASE TYPE 1) | ||||||
 キーワード キーワード |  TRANSFERASE (転移酵素) / AMINOGLYCOSIDE 6'-N-ACETYLTRANSFERASE / ANTIBIOTIC RESISTANCE (抗微生物薬耐性) / ACETYL COENZYME A (アセチルCoA) TRANSFERASE (転移酵素) / AMINOGLYCOSIDE 6'-N-ACETYLTRANSFERASE / ANTIBIOTIC RESISTANCE (抗微生物薬耐性) / ACETYL COENZYME A (アセチルCoA) | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 | ||||||
| 生物種 |  Enterococcus faecium (バクテリア) Enterococcus faecium (バクテリア) | ||||||
| 手法 | X線回折 / シンクロトロン / 多波長異常分散 / 解像度: 2.7 Å | ||||||
 データ登録者 データ登録者 | Wybenga-Groot, L.E. / Berghuis, A.M. | ||||||
 引用 引用 | ジャーナル: Structure Fold.Des. / 年: 1999 タイトル: Crystal structure of an aminoglycoside 6'-N-acetyltransferase: defining the GCN5-related N-acetyltransferase superfamily fold. 著者: Wybenga-Groot, L.E. / Draker, K. / Wright, G.D. / Berghuis, A.M. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1b87.cif.gz | 49.3 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1b87.ent.gz | 36.1 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1b87.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/b8/1b87ftp://data.pdbj.org/pub/pdb/validation_reports/b8/1b87 | HTTPS FTP |
|---|
-関連構造データ









-リンク
 PDBj
PDBj

- 集合体
集合体
| 登録構造単位 | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| 2 | 
| ||||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 20602.066 Da / 分子数: 1 / 変異: VAL127GLU / 由来タイプ: 天然 / 由来: (天然) Enterococcus faecium (バクテリア) / 参照: UniProt: Q47764 |
|---|---|
| #2: 化合物 | ChemComp-ACO / アセチルCoA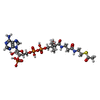  分子量: 809.571 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C23H38N7O17P3S 分子量: 809.571 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C23H38N7O17P3S |
| #3: 水 | ChemComp-HOH / 水 分子量: 18.015 Da / 分子数: 28 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 28 / 由来タイプ: 天然 / 式: H2O |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 3.2 Å3/Da / 溶媒含有率: 62 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | pH: 7.5 / 詳細: pH 7.5 | ||||||||||||||||||||||||||||||||||||
| 溶液の組成 |
| ||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 22 ℃ / 手法: 蒸気拡散法, ハンギングドロップ法 | ||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 103 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: CHESS  / ビームライン: F2 / 波長: 0.9809, 0.9795, 0.9792, 0.9770 / ビームライン: F2 / 波長: 0.9809, 0.9795, 0.9792, 0.9770 | |||||||||||||||
| 検出器 | タイプ: ADSC QUANTUM 4 / 検出器: CCD / 日付: 1998年1月15日 | |||||||||||||||
| 放射 | プロトコル: MAD / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray | |||||||||||||||
| 放射波長 |
| |||||||||||||||
| 反射 | 解像度: 2.8→40 Å / Num. obs: 6085 / % possible obs: 89.7 % / 冗長度: 2.1 % / Biso Wilson estimate: 19.2 Å2 / Rsym value: 12.7 / Net I/σ(I): 4.3 | |||||||||||||||
| 反射 シェル | 解像度: 2.8→2.94 Å / 冗長度: 2.1 % / Mean I/σ(I) obs: 1.4 / Rsym value: 33.2 / % possible all: 88.9 | |||||||||||||||
| 反射 | *PLUS Rmerge(I) obs: 0.127 | |||||||||||||||
| 反射 シェル | *PLUS % possible obs: 88.9 % / Rmerge(I) obs: 0.332 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多波長異常分散 / 解像度: 2.7→40 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 1 / 詳細: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 11.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.7→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.7→2.87 Å / Rfactor Rfree error: 0.027 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: X-PLOR / バージョン: 3.843 / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
