 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-8313: Structure of the SLC4 transporter Bor1p in an inward-facing confo... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8313 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the SLC4 transporter Bor1p in an inward-facing conformation | |||||||||||||||
 Map data Map data | Bor1p helical tube density map (Type 1) | |||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | boron transporter /  anion exchanger family / alternating access mechanism / Structural Genomics / PSI-Biology / Transcontinental EM Initiative for Membrane Protein Structure / TEMIMPS / TRANSPORT PROTEIN anion exchanger family / alternating access mechanism / Structural Genomics / PSI-Biology / Transcontinental EM Initiative for Membrane Protein Structure / TEMIMPS / TRANSPORT PROTEIN | |||||||||||||||
| Function / homology | Bicarbonate transporter, eukaryotic / Bicarbonate transporter-like, transmembrane domain / HCO3- transporter family / solute:inorganic anion antiporter activity / membrane => GO:0016020 / Bor1p boron transporter Function and homology information Function and homology information | |||||||||||||||
| Biological species |  Saccharomyces mikatae (yeast) Saccharomyces mikatae (yeast) | |||||||||||||||
| Method | helical reconstruction / cryo EM / Resolution: 5.9 Å | |||||||||||||||
 Authors Authors | Coudray N / Seyler S | |||||||||||||||
| Funding support |  United States, 4 items United States, 4 items
| |||||||||||||||
 Citation Citation | Journal: Protein Sci / Year: 2017 Title: Structure of the SLC4 transporter Bor1p in an inward-facing conformation. Authors: Nicolas Coudray / Sean L Seyler / Ralph Lasala / Zhening Zhang / Kathy M Clark / Mark E Dumont / Alexis Rohou / Oliver Beckstein / David L Stokes / Abstract: Bor1p is a secondary transporter in yeast that is responsible for boron transport. Bor1p belongs to the SLC4 family which controls bicarbonate exchange and pH regulation in animals as well as borate ...Bor1p is a secondary transporter in yeast that is responsible for boron transport. Bor1p belongs to the SLC4 family which controls bicarbonate exchange and pH regulation in animals as well as borate uptake in plants. The SLC4 family is more distantly related to members of the Amino acid-Polyamine-organoCation (APC) superfamily, which includes well studied transporters such as LeuT, Mhp1, AdiC, vSGLT, UraA, SLC26Dg. Their mechanism generally involves relative movements of two domains: a core domain that binds substrate and a gate domain that in many cases mediates dimerization. To shed light on conformational changes governing transport by the SLC4 family, we grew helical membrane crystals of Bor1p from Saccharomyces mikatae and determined a structure at ∼6 Å resolution using cryo-electron microscopy. To evaluate the conformation of Bor1p in these crystals, a homology model was built based on the related anion exchanger from red blood cells (AE1). This homology model was fitted to the cryo-EM density map using the Molecular Dynamics (MD) Flexible Fitting method and then relaxed by all-atom MD simulation in explicit solvent and membrane. Mapping of water accessibility indicates that the resulting structure represents an inward-facing conformation. Comparisons of the resulting Bor1p model with the X-ray structure of AE1 in an outward-facing conformation, together with MD simulations of inward-facing and outward-facing Bor1p models, suggest rigid body movements of the core domain relative to the gate domain. These movements are consistent with the rocking-bundle transport mechanism described for other members of the APC superfamily. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
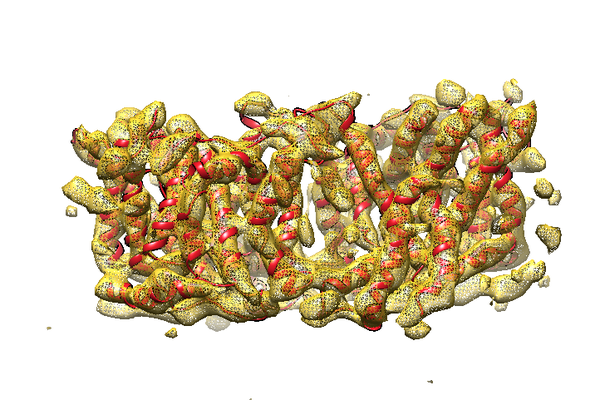
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8313.map.gz | 500 KB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8313-v30.xmlemd-8313.xml | 17.9 KB 17.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8313_fsc.xml | 4.2 KB | Display | FSC data file |
| Images | 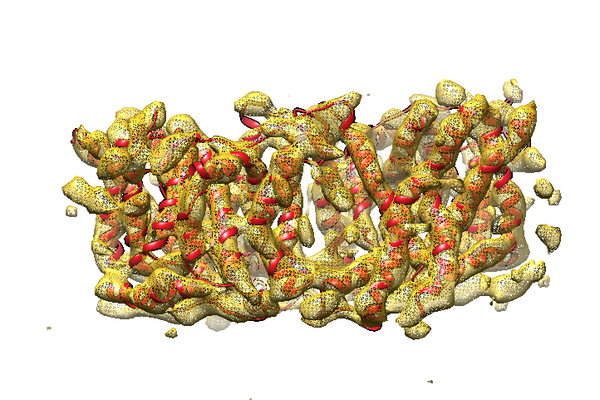 emd_8313.png emd_8313.png | 266.1 KB | ||
| Filedesc metadata | emd-8313.cif.gz | 6.9 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8313ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8313 http://ftp.pdbj.org/pub/emdb/structures/EMD-8313ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8313 | HTTPS FTP |
-Related structure data
| Related structure data |  5sv9MC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_8313.map.gz / Format: CCP4 / Size: 3.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Bor1p helical tube density map (Type 1) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.82 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Bor1p dimer in an inward-facing conformation
| Entire | Name: Bor1p dimer in an inward-facing conformation |
|---|---|
| Components |
|
-Supramolecule #1: Bor1p dimer in an inward-facing conformation
| Supramolecule | Name: Bor1p dimer in an inward-facing conformation / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Saccharomyces mikatae (yeast) |
| Molecular weight | Theoretical: 60 KDa |
-Macromolecule #1: Bor1p boron transporter
| Macromolecule | Name: Bor1p boron transporter / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Saccharomyces mikatae (yeast) |
| Molecular weight | Theoretical: 53.829766 KDa |
| Recombinant expression | Organism: Saccharomyces cerevisiae (brewer's yeast) |
| Sequence | String: IWLDLKDRIP YYKSDWVDAF NYRVIPSTVD TYFNNLLPAI AFAQDMFDRT DNSYGVNEVL LSSAMAGIVF GVLAGQPLCI VGVTGPISI FNYTVYEIIK PLNTSYFGFM FWICLWSMIF HLLLAFTNVV CLLQYVTTFP CDIFGLFINV VYIQKGIQIL T RQFHNTSG ...String: IWLDLKDRIP YYKSDWVDAF NYRVIPSTVD TYFNNLLPAI AFAQDMFDRT DNSYGVNEVL LSSAMAGIVF GVLAGQPLCI VGVTGPISI FNYTVYEIIK PLNTSYFGFM FWICLWSMIF HLLLAFTNVV CLLQYVTTFP CDIFGLFINV VYIQKGIQIL T RQFHNTSG EKSVQDGFAS VVVALVMTAF GLFFKSFHHY PLFTHKIRTF ISDYSTALSV LFWSSFTHFG GYLNDVKFKK LP ITKSFFP TSKFNRPQNT WLAYEPIPVK DVFIALPFGI ILTILFYFDH NVSSLMAQRH QYKLRKPSSF HYDFALLGLT TCI SGVLGI PAPNGLIPQA PLHTETLLVR DSNQNVVRCV EQRLTNTFQG LMILGTMTRP LLVCLGEIPQ AVLSGLFFIM GING LMTNV IIHRIVFLFS DPKRRDNNSP LAKISKRSMV IFLCFSLAGF TGEFAITNTI AAIGFPLVLL LSVIVSFSFT Y |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | helical reconstruction |
| Aggregation state | helical array |
-Sample preparation
| Concentration | 0.25 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7 Component:
Details: Buffer was changed twice per day. | |||||||||||||||
| Grid | Model: EMS / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. / Pretreatment - Atmosphere: OTHER | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Instrument: HOMEMADE PLUNGER | |||||||||||||||
| Details | The protein was solubilized in 1% n-Dodecyl beta-D-maltoside and exchanged into heptaethyleneglycol-n-dodecylether (C12E7) while bound to the IgG Sepharose affinity column. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Electron beam | Acceleration voltage: 200 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Calibrated defocus max: 2.7 µm / Calibrated defocus min: 0.85 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.0 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 19000 |
| Sample stage | Specimen holder model: GATAN 626 SINGLE TILT LIQUID NITROGEN CRYO TRANSFER HOLDER Cooling holder cryogen: NITROGEN |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Frames/image: 2-27 / Number grids imaged: 2 / Number real images: 252 / Average exposure time: 6.75 sec. / Average electron dose: 45.0 e/Å2 |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
-Image processing
| Segment selection | Number selected: 87 / Software - Name: SPARX/EMAN2 (ver. EMAN2.1) / Software - details: sxhelixboxer.py | ||||||
|---|---|---|---|---|---|---|---|
| Startup model | Type of model: OTHER Details: Initial model was created using the Fourier-Bessel method using 41 tube sections. | ||||||
| Final angle assignment | Type: NOT APPLICABLE / Software - Name: FREALIX (ver. 1.2.0) / Software - details: flx_wrap.rb | ||||||
| Final reconstruction | Applied symmetry - Helical parameters - Δz: 4.8 Å Applied symmetry - Helical parameters - Δ&Phi: 37.35 ° Applied symmetry - Helical parameters - Axial symmetry: C1 (asymmetric) Resolution.type: BY AUTHOR / Resolution: 5.9 Å / Resolution method: FSC 0.143 CUT-OFF Software:
Details: Reconstruction was done using D1 symmetry because of an existing two-fold symmetry in the unit cells composing the helical lattice. This two-fold axis runs perpendicular to the helical axis. ...Details: Reconstruction was done using D1 symmetry because of an existing two-fold symmetry in the unit cells composing the helical lattice. This two-fold axis runs perpendicular to the helical axis. For the final structure, a filter was applied to the map in order to compensate for resolution-dependent amplitude falloff. To do so, we built a model by arranging UraA in a helical assembly in order to mimic the mass distribution in Bor1p tubes. Fourier transforms from this model and from the experimental maps were then rotationally averaged to produce 1D scattering profiles. The resolution-dependent amplitude ratio from these profiles was used as a filter that was applied to the experimental amplitudes using SPARX routines. Finally, a low-pass filter was applied with a 5 Angstrom stop-band frequency. Number images used: 75 | ||||||
| FSC plot (resolution estimation) | 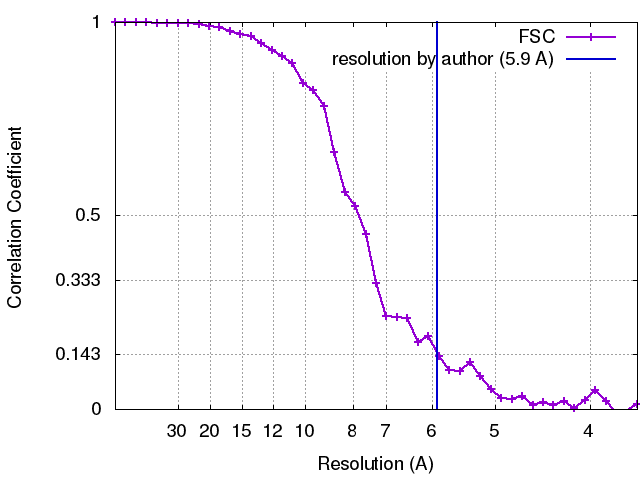 |
-Atomic model buiding 1
| Initial model |
| ||||||
|---|---|---|---|---|---|---|---|
| Details | A homology model was first created using human AE1 (PDB entry 4YZF) as a template using MODELLER, placed into the density map using SITUS, and then fitted into the map using the Molecular Dynamics Flexible Fitting (MDFF) method. | ||||||
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: cross-correlation coefficient | ||||||
| Output model | PDB-5sv9: |

