 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CryoEM Structure of Computationally Designed Nanocage O32-ZL4 | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | O32-ZL4 /  DE NOVO PROTEIN DE NOVO PROTEIN | |||||||||
| Function / homology | KDPG/KHG aldolase / KDPG and KHG aldolase / Aldolase-type TIM barrel / lyase activity / 2-dehydro-3-deoxyphosphogluconate aldolase/4-hydroxy-2-oxoglutarate aldolase Function and homology information Function and homology information | |||||||||
| Biological species |   Thermotoga maritima (bacteria) / synthetic construct (others) Thermotoga maritima (bacteria) / synthetic construct (others) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.9 Å | |||||||||
 Authors Authors | Weidle C / Borst A | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Nat Mater / Year: 2023 Title: Accurate computational design of three-dimensional protein crystals. Authors: Zhe Li / Shunzhi Wang / Una Nattermann / Asim K Bera / Andrew J Borst / Muammer Y Yaman / Matthew J Bick / Erin C Yang / William Sheffler / Byeongdu Lee / Soenke Seifert / Greg L Hura / ...Authors: Zhe Li / Shunzhi Wang / Una Nattermann / Asim K Bera / Andrew J Borst / Muammer Y Yaman / Matthew J Bick / Erin C Yang / William Sheffler / Byeongdu Lee / Soenke Seifert / Greg L Hura / Hannah Nguyen / Alex Kang / Radhika Dalal / Joshua M Lubner / Yang Hsia / Hugh Haddox / Alexis Courbet / Quinton Dowling / Marcos Miranda / Andrew Favor / Ali Etemadi / Natasha I Edman / Wei Yang / Connor Weidle / Banumathi Sankaran / Babak Negahdari / Michael B Ross / David S Ginger / David Baker /  Abstract: Protein crystallization plays a central role in structural biology. Despite this, the process of crystallization remains poorly understood and highly empirical, with crystal contacts, lattice packing ...Protein crystallization plays a central role in structural biology. Despite this, the process of crystallization remains poorly understood and highly empirical, with crystal contacts, lattice packing arrangements and space group preferences being largely unpredictable. Programming protein crystallization through precisely engineered side-chain-side-chain interactions across protein-protein interfaces is an outstanding challenge. Here we develop a general computational approach for designing three-dimensional protein crystals with prespecified lattice architectures at atomic accuracy that hierarchically constrains the overall number of degrees of freedom of the system. We design three pairs of oligomers that can be individually purified, and upon mixing, spontaneously self-assemble into >100 µm three-dimensional crystals. The structures of these crystals are nearly identical to the computational design models, closely corresponding in both overall architecture and the specific protein-protein interactions. The dimensions of the crystal unit cell can be systematically redesigned while retaining the space group symmetry and overall architecture, and the crystals are extremely porous and highly stable. Our approach enables the computational design of protein crystals with high accuracy, and the designed protein crystals, which have both structural and assembly information encoded in their primary sequences, provide a powerful platform for biological materials engineering. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_40926.map.gz | 266.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-40926-v30.xmlemd-40926.xml | 18.4 KB 18.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_40926_fsc.xml | 13.9 KB | Display | FSC data file |
| Images |  emd_40926.png emd_40926.png | 107.3 KB | ||
| Filedesc metadata | emd-40926.cif.gz | 6.4 KB | ||
| Others | emd_40926_half_map_1.map.gzemd_40926_half_map_2.map.gz | 261.6 MB 261.6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-40926ftp://ftp.pdbj.org/pub/emdb/structures/EMD-40926 http://ftp.pdbj.org/pub/emdb/structures/EMD-40926ftp://ftp.pdbj.org/pub/emdb/structures/EMD-40926 | HTTPS FTP |
-Related structure data
| Related structure data |  8szzMC  8cusC  8cutC  8cuuC  8cuvC  8cuwC  8cuxC  8cwsC  8cwyC  8cwzC  8farC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_40926.map.gz / Format: CCP4 / Size: 282.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Voxel size | X=Y=Z: 0.84 Å | ||||||||||||||||||||
| Density |
| ||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||
| Details | EMDB XML:
|
-Supplemental data
-Half map: #2
| File | emd_40926_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
 Z
Z Y
Y X
X








-Half map: #1
| File | emd_40926_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : O32-ZL4
| Entire | Name: O32-ZL4 |
|---|---|
| Components |
|
-Supramolecule #1: O32-ZL4
| Supramolecule | Name: O32-ZL4 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 Details: These two proteins were co expressed from the same plasmid and assembled inside E. coli into cages. O32-ZL4 was purified using the His tag on B component |
|---|---|
| Source (natural) | Organism: Thermotoga maritima (bacteria) |
| Molecular weight | Theoretical: 764.91792 KDa |
-Macromolecule #1: O32-ZL4 Component A
| Macromolecule | Name: O32-ZL4 Component A / type: protein_or_peptide / ID: 1 / Number of copies: 24 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 8.766459 KDa |
| Sequence | String: MTDELLRLAK EQAELLKEIK ILVELIAMLV KVIQKDPSDE ALKALAELVR KLKELVEDME RSMKEQLYII KGSWSG |
-Macromolecule #2: O32-ZL4 Component B
| Macromolecule | Name: O32-ZL4 Component B / type: protein_or_peptide / ID: 2 / Number of copies: 24 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Thermotoga maritima (bacteria) |
| Molecular weight | Theoretical: 23.170152 KDa |
| Recombinant expression | Organism: Escherichia coli BL21(DE3) (bacteria) |
| Sequence | String: MKMEELFKKH KIVAVLRAND AQEAREKALA VFEGGVHLIE ITFTVPNAAA VILLLSFLKE KGAIIGAGTV TSEEQCALAV LSGAEFIVS PHLDEEISQF CKEKGVFYMP GVMTPTELVK AMKLGHTILK LFPGEVVGPQ FVKAMKGPFP NVKFVPTGGV N LDNVCEWF ...String: MKMEELFKKH KIVAVLRAND AQEAREKALA VFEGGVHLIE ITFTVPNAAA VILLLSFLKE KGAIIGAGTV TSEEQCALAV LSGAEFIVS PHLDEEISQF CKEKGVFYMP GVMTPTELVK AMKLGHTILK LFPGEVVGPQ FVKAMKGPFP NVKFVPTGGV N LDNVCEWF KAGVLAVGVG SALVKGTPDE VREKAKAFVE KIRGCTELEH HHHHH UniProtKB: 2-dehydro-3-deoxyphosphogluconate aldolase/4-hydroxy-2-oxoglutarate aldolase |
-Macromolecule #3: SODIUM ION
| Macromolecule | Name: SODIUM ION / type: ligand / ID: 3 / Number of copies: 24 |
|---|---|
| Molecular weight | Theoretical: 22.99 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 2.1 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
Details: 25 mM Tris/HCl pH 7.5, 150 mM NaCl | |||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Support film - Film thickness: 2 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. / Pretreatment - Pressure: 0.0001 kPa | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 295 K / Instrument: FEI VITROBOT MARK IV | |||||||||
| Details | 25 mM Tris, 150 mM NaCl |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Nominal defocus max: 1.8 µm / Nominal defocus min: 0.8 µm |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 60.0 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 673044 |
|---|---|
| Startup model | Type of model: INSILICO MODEL |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD |
| Final 3D classification | Number classes: 1 |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD |
| Final reconstruction | Number classes used: 1 / Applied symmetry - Point group: O (octahedral) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 2.9 Å / Resolution method: FSC 0.143 CUT-OFF / Number images used: 674044 |
| FSC plot (resolution estimation) | 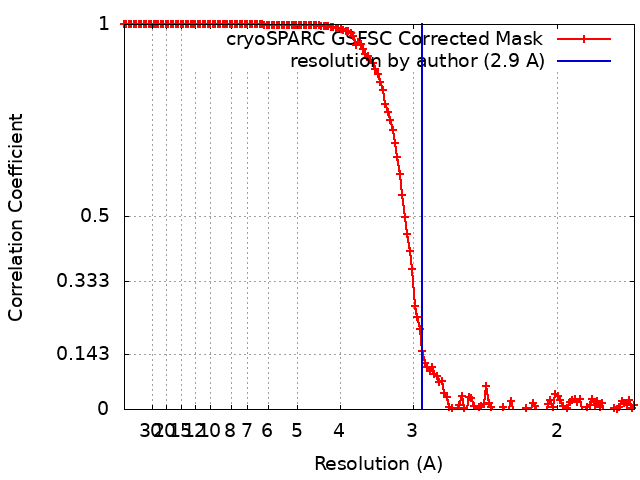 |
-Atomic model buiding 1
| Initial model | Chain - Source name: Other / Chain - Initial model type: in silico model / Details: Computational model |
|---|---|
| Refinement | Space: REAL / Protocol: AB INITIO MODEL / Overall B value: 144.9 / Target criteria: Cross-correlation coefficient |
| Output model | PDB-8szz: |
