 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3441: Subtomogram average of the mitochondrial ATP synthase dimer from ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3441 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
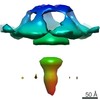
| Title | Subtomogram average of the mitochondrial ATP synthase dimer from the ciliate Paramecium tetraurelia | |||||||||
 Map data Map data | Subtomogram average of mitochondrial ATP synthase dimer from the ciliate Paramecium tetraurelia | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | ATP synthase dimers /  mitochondria / ciliate mitochondria / ciliate | |||||||||
| Biological species |  | |||||||||
| Method | electron tomography / cryo EM / Resolution: 26.0 Å | |||||||||
 Authors Authors | Muehleip AW / Kuehlbrandt W / Davies KM | |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2016 Title: Helical arrays of U-shaped ATP synthase dimers form tubular cristae in ciliate mitochondria. Authors: Alexander W Mühleip / Friederike Joos / Christoph Wigge / Achilleas S Frangakis / Werner Kühlbrandt / Karen M Davies /  Abstract: F1Fo-ATP synthases are universal energy-converting membrane protein complexes that synthesize ATP from ADP and inorganic phosphate. In mitochondria of yeast and mammals, the ATP synthase forms V- ...F1Fo-ATP synthases are universal energy-converting membrane protein complexes that synthesize ATP from ADP and inorganic phosphate. In mitochondria of yeast and mammals, the ATP synthase forms V-shaped dimers, which assemble into rows along the highly curved ridges of lamellar cristae. Using electron cryotomography and subtomogram averaging, we have determined the in situ structure and organization of the mitochondrial ATP synthase dimer of the ciliate Paramecium tetraurelia. The ATP synthase forms U-shaped dimers with parallel monomers. Each complex has a prominent intracrista domain, which links the c-ring of one monomer to the peripheral stalk of the other. Close interaction of intracrista domains in adjacent dimers results in the formation of helical ATP synthase dimer arrays, which differ from the loose dimer rows in all other organisms observed so far. The parameters of the helical arrays match those of the cristae tubes, suggesting the unique features of the P. tetraurelia ATP synthase are directly responsible for generating the helical tubular cristae. We conclude that despite major structural differences between ATP synthase dimers of ciliates and other eukaryotes, the formation of ATP synthase dimer rows is a universal feature of mitochondria and a fundamental determinant of cristae morphology. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3441.map.gz | 7.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3441-v30.xmlemd-3441.xml | 13.1 KB 13.1 KB | Display Display | EMDB header |
| Images | emd_3441.tif | 130.7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3441ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3441 http://ftp.pdbj.org/pub/emdb/structures/EMD-3441ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3441 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|





-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_3441.map.gz / Format: CCP4 / Size: 10.2 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Subtomogram average of mitochondrial ATP synthase dimer from the ciliate Paramecium tetraurelia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 4.46 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Mitochondrial ATP synthase dimer from Paramecium tetraurelia
| Entire | Name: Mitochondrial ATP synthase dimer from Paramecium tetraurelia |
|---|---|
| Components |
|
-Supramolecule #1000: Mitochondrial ATP synthase dimer from Paramecium tetraurelia
| Supramolecule | Name: Mitochondrial ATP synthase dimer from Paramecium tetraurelia type: sample / ID: 1000 / Oligomeric state: dimer / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 1.2 MDa |
-Macromolecule #1: Mitochondrial F1FoATPase
| Macromolecule | Name: Mitochondrial F1FoATPase / type: protein_or_peptide / ID: 1 / Name.synonym: Mitochondrial ATP synthase dimer / Number of copies: 1 / Oligomeric state: Dimer / Recombinant expression: No / Database: NCBI |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 1.2 MDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | electron tomography |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.4 / Details: 20 mM Tris, 250 mM trehalose |
|---|---|
| Grid | Details: 300 mesh Quantifoil copper grid R2/2 |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 10 % / Chamber temperature: 110 K / Instrument: OTHER Method: manual blotting for 5-6 seconds with Whatman paper #4 |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 4.0 µm / Nominal defocus min: 2.5 µm / Nominal magnification: 64000 |
| Specialist optics | Energy filter - Name: GIF Quantum, Gatan / Energy filter - Lower energy threshold: 0.0 eV / Energy filter - Upper energy threshold: 20.0 eV |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Tilt series - Axis1 - Min angle: -60 ° / Tilt series - Axis1 - Max angle: 60 ° / Tilt series - Axis1 - Angle increment: 2 ° |
| Temperature | Min: 80 K |
| Alignment procedure | Legacy - Astigmatism: Objective lens astigmatism was corrected at 64,000 times magnification |
| Date | Jul 3, 2013 |
| Image recording | Category: CCD / Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Number real images: 720 / Average electron dose: 100 e/Å2 / Camera length: 19.3 / Bits/pixel: 32 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| CTF correction | Details: each projection, strip-based approach |
|---|---|
| Final reconstruction | Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 26.0 Å / Resolution method: OTHER / Software - Name: IMOD, PEET Details: Final subtomogram average was generated from 1244 subvolumes. Number images used: 60 |
| Details | IMOD was used to align tilt series based on the position of gold fiducials, correct CTF on each projection image, and generate a tomographic volume using weighted back-projection. Subtomogram averaging was performed with PEET after initial estimate of rotations were calculated based on the orientation of particles relative to the membrane. |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C / Chain - #3 - Chain ID: D / Chain - #4 - Chain ID: E / Chain - #5 - Chain ID: F / Chain - #6 - Chain ID: G / Chain - #7 - Chain ID: H / Chain - #8 - Chain ID: I |
|---|---|
| Software | Name: Chimera |
| Refinement | Space: RECIPROCAL / Protocol: RIGID BODY FIT / Target criteria: cross correlation coefficient maximization |

-Atomic model buiding 2
| Initial model | PDB ID: Chain - #0 - Chain ID: S / Chain - #1 - Chain ID: W |
|---|---|
| Software | Name: Chimera |
| Refinement | Space: RECIPROCAL / Protocol: RIGID BODY FIT / Target criteria: cross correlation coefficient maximization |

