 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-33464: Structure of SARS-Cov-2 D614G spike protein (single Arg S1/S2 cle... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of SARS-Cov-2 D614G spike protein (single Arg S1/S2 cleavage site), Open Conformation | ||||||||||||
 Map data Map data | |||||||||||||
 Sample Sample |
| ||||||||||||
| Biological species |    Severe acute respiratory syndrome coronavirus 2 Severe acute respiratory syndrome coronavirus 2 | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.1 Å | ||||||||||||
 Authors Authors | Qu K / Chen Q / Ciazynska KA / Liu B / Zhang X / Wang J / He Y / Guan J / He J / Liu T ...Qu K / Chen Q / Ciazynska KA / Liu B / Zhang X / Wang J / He Y / Guan J / He J / Liu T / Carter AP / Xiong X / Briggs JAG | ||||||||||||
| Funding support | European Union,  United Kingdom, 3 items United Kingdom, 3 items
| ||||||||||||
 Citation Citation | Journal: PLoS Pathog / Year: 2022 Title: Engineered disulfide reveals structural dynamics of locked SARS-CoV-2 spike. Authors: Kun Qu / Qiuluan Chen / Katarzyna A Ciazynska / Banghui Liu / Xixi Zhang / Jingjing Wang / Yujie He / Jiali Guan / Jun He / Tian Liu / Xiaofei Zhang / Andrew P Carter / Xiaoli Xiong / John A G Briggs /    Abstract: The spike (S) protein of SARS-CoV-2 has been observed in three distinct pre-fusion conformations: locked, closed and open. Of these, the function of the locked conformation remains poorly understood. ...The spike (S) protein of SARS-CoV-2 has been observed in three distinct pre-fusion conformations: locked, closed and open. Of these, the function of the locked conformation remains poorly understood. Here we engineered a SARS-CoV-2 S protein construct "S-R/x3" to arrest SARS-CoV-2 spikes in the locked conformation by a disulfide bond. Using this construct we determined high-resolution structures confirming that the x3 disulfide bond has the ability to stabilize the otherwise transient locked conformations. Structural analyses reveal that wild-type SARS-CoV-2 spike can adopt two distinct locked-1 and locked-2 conformations. For the D614G spike, based on which all variants of concern were evolved, only the locked-2 conformation was observed. Analysis of the structures suggests that rigidified domain D in the locked conformations interacts with the hinge to domain C and thereby restrains RBD movement. Structural change in domain D correlates with spike conformational change. We propose that the locked-1 and locked-2 conformations of S are present in the acidic high-lipid cellular compartments during virus assembly and egress. In this model, release of the virion into the neutral pH extracellular space would favour transition to the closed or open conformations. The dynamics of this transition can be altered by mutations that modulate domain D structure, as is the case for the D614G mutation, leading to changes in viral fitness. The S-R/x3 construct provides a tool for the further structural and functional characterization of the locked conformations of S, as well as how sequence changes might alter S assembly and regulation of receptor binding domain dynamics. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_33464.map.gz | 106.2 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-33464-v30.xmlemd-33464.xml | 19 KB 19 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_33464_fsc.xml | 12.7 KB | Display | FSC data file |
| Images | 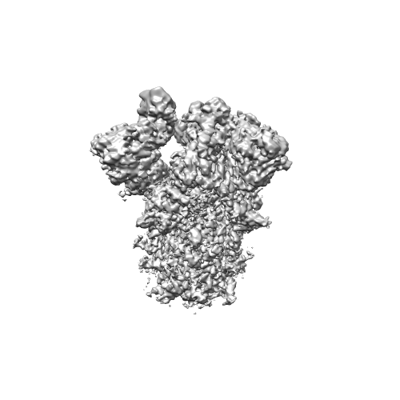 emd_33464.png emd_33464.png | 58.4 KB | ||
| Others | emd_33464_half_map_1.map.gzemd_33464_half_map_2.map.gz | 140.6 MB 140.9 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-33464ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33464 http://ftp.pdbj.org/pub/emdb/structures/EMD-33464ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33464 | HTTPS FTP |
-Related structure data



















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_33464.map.gz / Format: CCP4 / Size: 178 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Voxel size | X=Y=Z: 1.061 Å | ||||||||||||||||||||
| Density |
| ||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||
| Details | EMDB XML:
|
-Supplemental data
-Half map: #1
| File | emd_33464_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
 Z
Z Y
Y X
X









-Half map: #2
| File | emd_33464_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Severe acute respiratory syndrome coronavirus 2 Spike protein
| Entire | Name: Severe acute respiratory syndrome coronavirus 2 Spike protein |
|---|---|
| Components |
|
-Supramolecule #1: Severe acute respiratory syndrome coronavirus 2 Spike protein
| Supramolecule | Name: Severe acute respiratory syndrome coronavirus 2 Spike protein type: complex / Chimera: Yes / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Severe acute respiratory syndrome coronavirus 2 |
| Recombinant expression | Organism:  Homo sapiens (human) Homo sapiens (human) |
-Macromolecule #1: Spike glycoprotein
| Macromolecule | Name: Spike glycoprotein / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Severe acute respiratory syndrome coronavirus 2 |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: ETGTQCVNLT TRTQLPPAYT NSFTRGVYYP DKVFRSSVLH STQDLFLPFF SNVTWFHAIH VSGTNGTKRF DNPVLPFNDG VYFASTEKSN IIRGWIFGTT LDSKTQSLLI VNNATNVVIK VCEFQFCNDP FLGVYYHKNN KSWMESEFRV YSSANNCTFE YVSQPFLMDL ...String: ETGTQCVNLT TRTQLPPAYT NSFTRGVYYP DKVFRSSVLH STQDLFLPFF SNVTWFHAIH VSGTNGTKRF DNPVLPFNDG VYFASTEKSN IIRGWIFGTT LDSKTQSLLI VNNATNVVIK VCEFQFCNDP FLGVYYHKNN KSWMESEFRV YSSANNCTFE YVSQPFLMDL EGKQGNFKNL REFVFKNIDG YFKIYSKHTP INLVRDLPQG FSALEPLVDL PIGINITRFQ TLLALHRSYL TPGDSSSGWT AGAAAYYVGY LQPRTFLLKY NENGTITDAV DCALDPLSET KCTLKSFTVE KGIYQTSNFR VQPTESIVRF PNITNLCPFG EVFNATRFAS VYAWNRKRIS NCVADYSVLY NSASFSTFKC YGVSPTKLND LCFTNVYADS FVIRGDEVRQ IAPGQTGKIA DYNYKLPDDF TGCVIAWNSN NLDSKVGGNY NYLYRLFRKS NLKPFERDIS TEIYQAGSTP CNGVEGFNCY FPLQSYGFQP TNGVGYQPYR VVVLSFELLH APATVCGPKK STNLVKNKCV NFNFNGLTGT GVLTESNKKF LPFQQFGRDI ADTTDAVRDP QTLEILDITP CSFGGVSVIT PGTNTSNQVA VLYQGVNCTE VPVAIHADQL TPTWRVYSTG SNVFQTRAGC LIGAEHVNNS YECDIPIGAG ICASYQTQTN SRSVASQSII AYTMSLGAEN SVAYSNNSIA IPTNFTISVT TEILPVSMTK TSVDCTMYIC GDSTECSNLL LQYGSFCTQL NRALTGIAVE QDKNTQEVFA QVKQIYKTPP IKDFGGFNFS QILPDPSKPS KRSFIEDLLF NKVTLADAGF IKQYGDCLGD IAARDLICAQ KFNGLTVLPP LLTDEMIAQY TSALLAGTIT SGWTFGAGAA LQIPFAMQMA YRFNGIGVTQ NVLYENQKLI ANQFNSAIGK IQDSLSSTAS ALGKLQDVVN QNAQALNTLV KQLSSNFGAI SSVLNDILSR LDKVEAEVQI DRLITGRLQS LQTYVTQQLI RAAEIRASAN LAATKMSECV LGQSKRVDFC GKGYHLMSFP QSAPHGVVFL HVTYVPAQEK NFTTAPAICH DGKAHFPREG VFVSNGTHWF VTQRNFYEPQ IITTDNTFVS GNCDVVIGIV NNTVYDPLQP ELDSFKEELD KYFKNHTSPD VDLGDISGIN ASVVNIQKEI DRLNEVAKNL NESLIDLQEL GKYEQYIKGS GRENLYFQGG GGSGYIPEAP RDGQAYVRKD GEWVLLSTFL GHHHHHH |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.6 mg/mL |
|---|---|
| Buffer | pH: 7.4 |
| Grid | Model: C-flat-2/2 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 2.4 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 81000 |
| Specialist optics | Energy filter - Name: GIF Quantum LS |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Number grids imaged: 1 / Number real images: 1980 / Average electron dose: 50.0 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 1728058 / Details: Template picking |
|---|---|
| CTF correction | Software - Name: CTFFIND (ver. 4.1.13) |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) |
| Final 3D classification | Number classes: 6 / Software - Name: RELION (ver. 3.1) |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) |
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 3.1 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 3.1) / Number images used: 284090 |
| FSC plot (resolution estimation) |  |
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: Correlation coefficient |
|---|
