 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-1998 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | ATP-triggered molecular mechanics of the chaperonin GroEL | |||||||||
 マップデータ マップデータ | The GroEL-ATP7 Rs1 map is one of seven maps calculated from a heterogenous sample | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | Tetradecamer of GroEL with ATP bound in one ring | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報GroEL-GroES complex / chaperonin ATPase / virion assembly / : / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / protein folding / response to heat ...GroEL-GroES complex / chaperonin ATPase / virion assembly / : / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / protein folding / response to heat / protein refolding / magnesium ion binding / ATP hydrolysis activity / ATP binding / identical protein binding / membrane / cytosol 類似検索 - 分子機能 | |||||||||
| 生物種 |  | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 8.0 Å | |||||||||
 データ登録者 データ登録者 | Clare DK / Vasishtan D / Stagg S / Quispe J / Farr GW / Topf M / Horwich AL / Saibil HR | |||||||||
 引用 引用 | ジャーナル: Cell / 年: 2012 タイトル: ATP-triggered conformational changes delineate substrate-binding and -folding mechanics of the GroEL chaperonin. 著者: Daniel K Clare / Daven Vasishtan / Scott Stagg / Joel Quispe / George W Farr / Maya Topf / Arthur L Horwich / Helen R Saibil /  要旨: The chaperonin GroEL assists the folding of nascent or stress-denatured polypeptides by actions of binding and encapsulation. ATP binding initiates a series of conformational changes triggering the ...The chaperonin GroEL assists the folding of nascent or stress-denatured polypeptides by actions of binding and encapsulation. ATP binding initiates a series of conformational changes triggering the association of the cochaperonin GroES, followed by further large movements that eject the substrate polypeptide from hydrophobic binding sites into a GroES-capped, hydrophilic folding chamber. We used cryo-electron microscopy, statistical analysis, and flexible fitting to resolve a set of distinct GroEL-ATP conformations that can be ordered into a trajectory of domain rotation and elevation. The initial conformations are likely to be the ones that capture polypeptide substrate. Then the binding domains extend radially to separate from each other but maintain their binding surfaces facing the cavity, potentially exerting mechanical force upon kinetically trapped, misfolded substrates. The extended conformation also provides a potential docking site for GroES, to trigger the final, 100° domain rotation constituting the "power stroke" that ejects substrate into the folding chamber. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
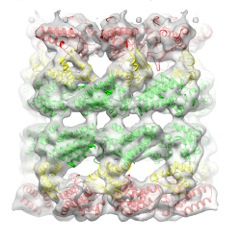
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_1998.map.gz | 481.6 KB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-1998-v30.xmlemd-1998.xml | 11.1 KB 11.1 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  EMD1998.jpeg EMD1998.jpeg | 27.6 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1998ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1998 http://ftp.pdbj.org/pub/emdb/structures/EMD-1998ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1998 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_1998_validation.pdf.gz | 214.9 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_1998_full_validation.pdf.gz | 214 KB | 表示 | |
| XML形式データ | emd_1998_validation.xml.gz | 5.9 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1998ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1998 | HTTPS FTP |
-関連構造データ
| 関連構造データ |  4aaqMC  1997C  1999C  2000C  2001C  2002C  2003C  4aarC  4aasC  4aauC  4ab2C  4ab3C M: このマップから作成された原子モデル C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ |






-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |

-マップ
| ファイル | ダウンロード / ファイル: emd_1998.map.gz / 形式: CCP4 / 大きさ: 26.4 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | The GroEL-ATP7 Rs1 map is one of seven maps calculated from a heterogenous sample | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 2.02 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : GroEL-ATP7 Rs1
| 全体 | 名称: GroEL-ATP7 Rs1 |
|---|---|
| 要素 |
|
-超分子 #1000: GroEL-ATP7 Rs1
| 超分子 | 名称: GroEL-ATP7 Rs1 / タイプ: sample / ID: 1000 集合状態: tetradecamer of GroEL with 7 ATP molecules bound Number unique components: 2 |
|---|---|
| 分子量 | 実験値: 800 KDa / 理論値: 800 KDa |
-分子 #1: hsp60
| 分子 | 名称: hsp60 / タイプ: protein_or_peptide / ID: 1 / Name.synonym: GroEL / 詳細: ATPase Mutant, D398A / コピー数: 14 / 集合状態: tetradecamer / 組換発現: Yes |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 実験値: 56 KDa / 理論値: 56 KDa |
| 組換発現 | 生物種: |
| 配列 | GO: protein refolding / InterPro: Chaperonin Cpn60/GroEL/TCP-1 family |
-分子 #2: ATP
| 分子 | 名称: ATP / タイプ: ligand / ID: 2 / Name.synonym: ATP / 詳細: ATP is bound to seven subunits of one ring / 組換発現: No |
|---|---|
| 由来(天然) | 生物種: synthetic construct (人工物) |
| 分子量 | 実験値: 550 Da / 理論値: 550 Da |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 4 mg/mL |
|---|---|
| 緩衝液 | pH: 7.4 詳細: 50 mM Tris-HCl pH 7.4, 50 mM KCl and 10 mM MgCl2, 200uM ATP |
| グリッド | 詳細: cflat grids r2/2 |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 95 K / 装置: OTHER / 詳細: Vitrification instrument: Vitrobot / Timed resolved state: Vitrified within 30 seconds / 手法: Grids were blotted for 2-3 seconds |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TECNAI F20 |
|---|---|
| 温度 | 最低: 95 K / 最高: 95 K / 平均: 95 K |
| 詳細 | The data were collected with Leginon at SCRIPPS |
| 撮影 | カテゴリ: CCD フィルム・検出器のモデル: GATAN ULTRASCAN 4000 (4k x 4k) 平均電子線量: 15 e/Å2 |
| 電子線 | 加速電圧: 120 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 倍率(補正後): 148500 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2 mm / 最大 デフォーカス(公称値): 3.5 µm / 最小 デフォーカス(公称値): 0.7 µm |
| 試料ステージ | 試料ホルダー: Eucentric / 試料ホルダーモデル: GATAN LIQUID NITROGEN |
| 実験機器 |  モデル: Tecnai F20 / 画像提供: FEI Company |
-画像解析
| 詳細 | The particles were automatically picked using FindEM |
|---|---|
| CTF補正 | 詳細: Each particle was phase flipped |
| 最終 再構成 | 想定した対称性 - 点群: C7 (7回回転対称) / アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 8.0 Å / 解像度の算出法: FSC 0.5 CUT-OFF / ソフトウェア - 名称: SPIDER, IMAGIC / 詳細: SIRT was used to reconstruct the final map / 使用した粒子像数: 5500 |
| 最終 角度割当 | 詳細: theta 80-100, phi 0-51.42 |

