Netherlands Organisation for Scientific Research (NWO)
VI.Veni.202.271
Netherlands
Chinese Scholarship Council
2014-03250042
China
Citation
Journal: Nature / Year: 2023 Title: Sialoglycan binding triggers spike opening in a human coronavirus. Authors: Matti F Pronker / Robert Creutznacher / Ieva Drulyte / Ruben J G Hulswit / Zeshi Li / Frank J M van Kuppeveld / Joost Snijder / Yifei Lang / Berend-Jan Bosch / Geert-Jan Boons / Martin Frank ...Authors: Matti F Pronker / Robert Creutznacher / Ieva Drulyte / Ruben J G Hulswit / Zeshi Li / Frank J M van Kuppeveld / Joost Snijder / Yifei Lang / Berend-Jan Bosch / Geert-Jan Boons / Martin Frank / Raoul J de Groot / Daniel L Hurdiss / Abstract: Coronavirus spike proteins mediate receptor binding and membrane fusion, making them prime targets for neutralizing antibodies. In the cases of severe acute respiratory syndrome coronavirus, severe ...Coronavirus spike proteins mediate receptor binding and membrane fusion, making them prime targets for neutralizing antibodies. In the cases of severe acute respiratory syndrome coronavirus, severe acute respiratory syndrome coronavirus 2 and Middle East respiratory syndrome coronavirus, spike proteins transition freely between open and closed conformations to balance host cell attachment and immune evasion. Spike opening exposes domain S1, allowing it to bind to proteinaceous receptors, and is also thought to enable protein refolding during membrane fusion. However, with a single exception, the pre-fusion spike proteins of all other coronaviruses studied so far have been observed exclusively in the closed state. This raises the possibility of regulation, with spike proteins more commonly transitioning to open states in response to specific cues, rather than spontaneously. Here, using cryogenic electron microscopy and molecular dynamics simulations, we show that the spike protein of the common cold human coronavirus HKU1 undergoes local and long-range conformational changes after binding a sialoglycan-based primary receptor to domain S1. This binding triggers the transition of S1 domains to the open state through allosteric interdomain crosstalk. Our findings provide detailed insight into coronavirus attachment, with possibilities of dual receptor usage and priming of entry as a means of immune escape.
Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 1 / Number real images: 4207 / Average exposure time: 2.68 sec. / Average electron dose: 46.0 e/Å2
Experimental equipment
Model: Titan Krios / Image courtesy: FEI Company
-
Image processing
Particle selection
Number selected: 9144772
Startup model
Type of model: OTHER / Details: Ab initio
Initial angle assignment
Type: MAXIMUM LIKELIHOOD / Software - Name: cryoSPARC (ver. 4.1) / Details: Ab initio
Final angle assignment
Type: MAXIMUM LIKELIHOOD / Software - Name: cryoSPARC (ver. 4.1)
Final reconstruction
Number classes used: 1 / Applied symmetry - Point group: C3 (3 fold cyclic) / Resolution.type: BY AUTHOR / Resolution: 3.4 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: cryoSPARC (ver. 4.1) / Number images used: 108396
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information
 Map data
Map data Sample
Sample Keywords
Keywords Virus /
Virus /  Function and homology information
Function and homology information
 Authors
Authors Netherlands,
Netherlands,  China, 2 items
China, 2 items  Citation
Citation
 Structure visualization
Structure visualization
 Downloads & links
Downloads & links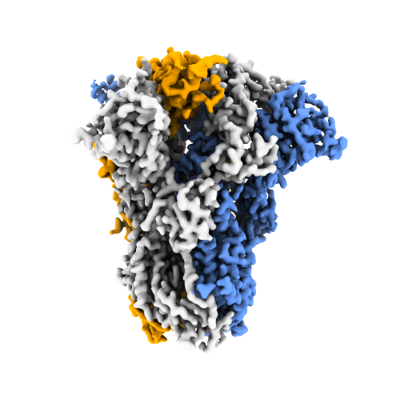 emd_16882.png
emd_16882.png http://ftp.pdbj.org/pub/emdb/structures/EMD-16882
http://ftp.pdbj.org/pub/emdb/structures/EMD-16882











 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















































 Sample components
Sample components

 Processing
Processing Electron microscopy
Electron microscopy


