 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-15903: Automated simulation-based refinement of maltoporin into a cryo-E... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Automated simulation-based refinement of maltoporin into a cryo-EM density | |||||||||
 Map data Map data | Half map 2, box 128 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords |  maltoporin / density fit / automated refinement / cryo-em / MEMBRANE PROTEIN maltoporin / density fit / automated refinement / cryo-em / MEMBRANE PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationmaltodextrin transmembrane transporter activity / maltose transporting porin activity / pore complex / monoatomic ion transport / cell outer membrane Similarity search - Function | |||||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.0 Å | |||||||||
 Authors Authors | Yvonnesdotter L / Rovsnik U / Blau C / Lycksell M / Howard RJ / Lindahl E | |||||||||
| Funding support |  Sweden, 2 items Sweden, 2 items
| |||||||||
 Citation Citation | Journal: Biophys J / Year: 2023 Title: Automated simulation-based membrane protein refinement into cryo-EM data. Authors: Linnea Yvonnesdotter / Urška Rovšnik / Christian Blau / Marie Lycksell / Rebecca Joy Howard / Erik Lindahl / Abstract: The resolution revolution has increasingly enabled single-particle cryogenic electron microscopy (cryo-EM) reconstructions of previously inaccessible systems, including membrane proteins-a category ...The resolution revolution has increasingly enabled single-particle cryogenic electron microscopy (cryo-EM) reconstructions of previously inaccessible systems, including membrane proteins-a category that constitutes a disproportionate share of drug targets. We present a protocol for using density-guided molecular dynamics simulations to automatically refine atomistic models into membrane protein cryo-EM maps. Using adaptive force density-guided simulations as implemented in the GROMACS molecular dynamics package, we show how automated model refinement of a membrane protein is achieved without the need to manually tune the fitting force ad hoc. We also present selection criteria to choose the best-fit model that balances stereochemistry and goodness of fit. The proposed protocol was used to refine models into a new cryo-EM density of the membrane protein maltoporin, either in a lipid bilayer or detergent micelle, and we found that results do not substantially differ from fitting in solution. Fitted structures satisfied classical model-quality metrics and improved the quality and the model-to-map correlation of the x-ray starting structure. Additionally, the density-guided fitting in combination with generalized orientation-dependent all-atom potential was used to correct the pixel-size estimation of the experimental cryo-EM density map. This work demonstrates the applicability of a straightforward automated approach to fitting membrane protein cryo-EM densities. Such computational approaches promise to facilitate rapid refinement of proteins under different conditions or with various ligands present, including targets in the highly relevant superfamily of membrane proteins. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_15903.map.gz | 7.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-15903-v30.xmlemd-15903.xml | 16 KB 16 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_15903_fsc.xml | 9.1 KB | Display | FSC data file |
| Images | 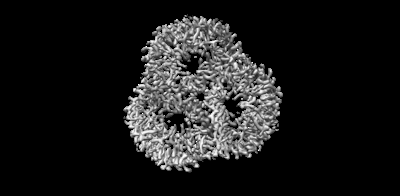 emd_15903.png emd_15903.png | 40.8 KB | ||
| Masks | emd_15903_msk_1.map | 8 MB | Mask map | |
| Others | emd_15903_additional_1.map.gzemd_15903_half_map_1.map.gzemd_15903_half_map_2.map.gz | 3.4 MB 7.4 MB 7.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-15903ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15903 http://ftp.pdbj.org/pub/emdb/structures/EMD-15903ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15903 | HTTPS FTP |
-Related structure data
| Related structure data |  8b7vMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_15903.map.gz / Format: CCP4 / Size: 8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
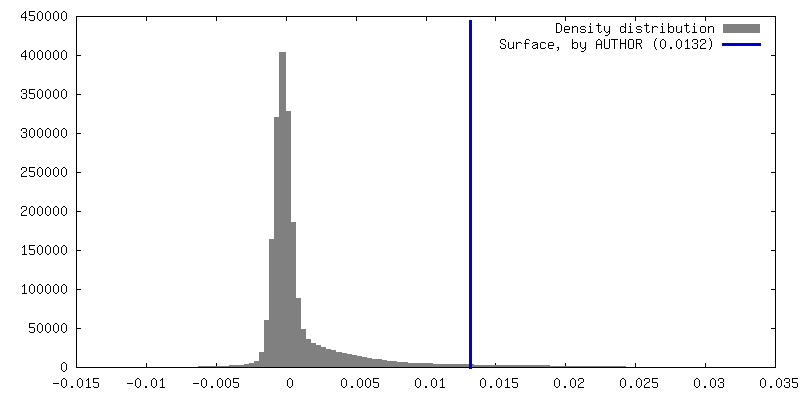
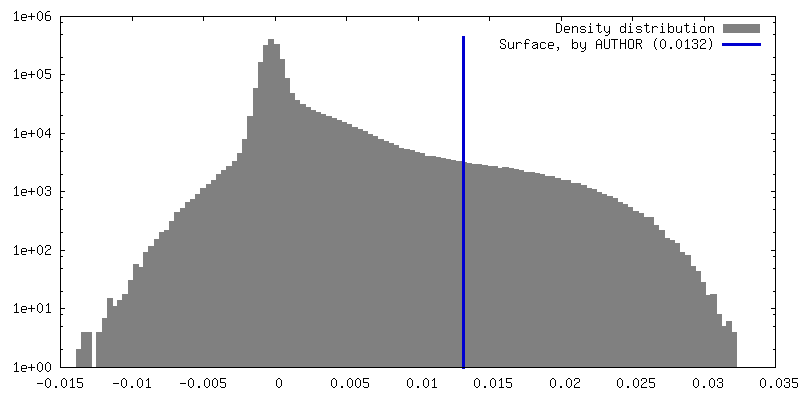
| Annotation | Half map 2, box 128 | ||||||||||||||||||||||||||||||||||||
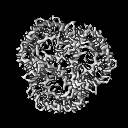
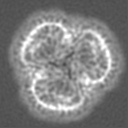
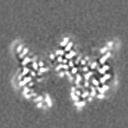
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.83 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)










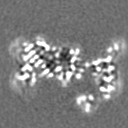
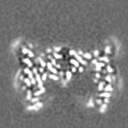

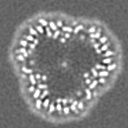

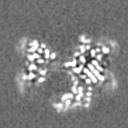
-Supplemental data
-Mask #1
| File | emd_15903_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
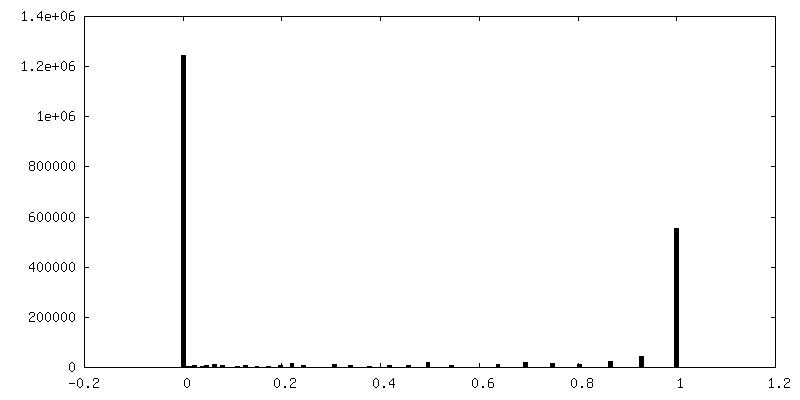
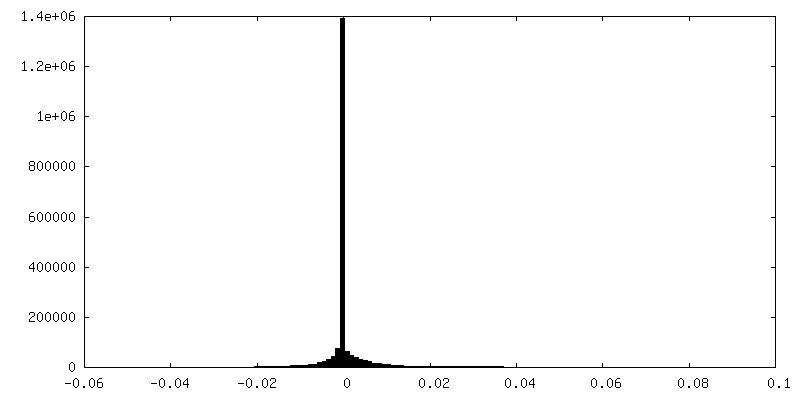
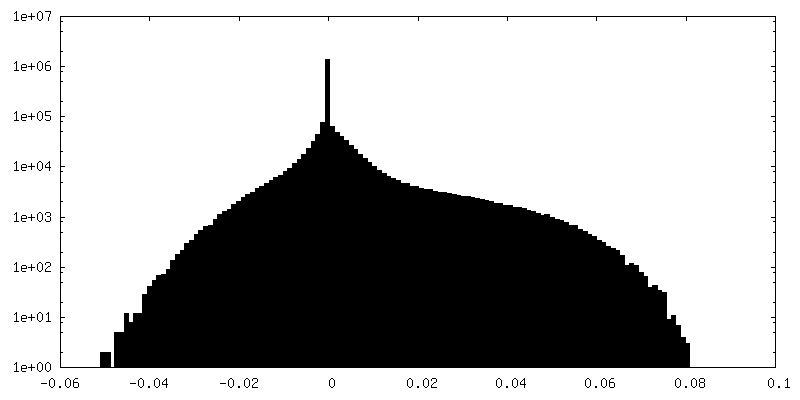
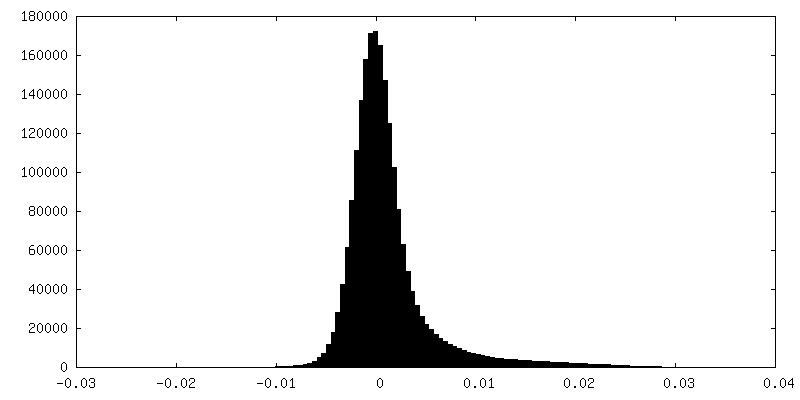
| Density Histograms |







-Additional map: Postprocessed sharpened map, box 128
| File | emd_15903_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Postprocessed sharpened map, box 128 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |





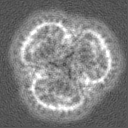
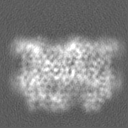
-Half map: 3D refined model used for automated fitting, box 128
| File | emd_15903_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 3D refined model used for automated fitting, box 128 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

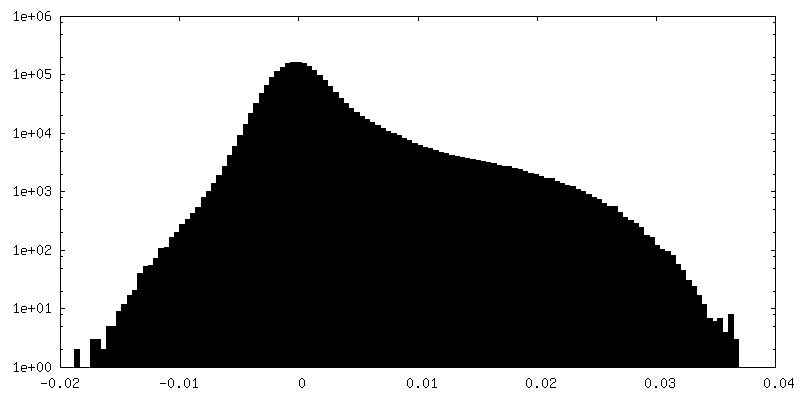
-Half map: Half map 1, box 128
| File | emd_15903_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 1, box 128 | ||||||||||||
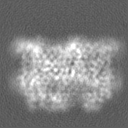
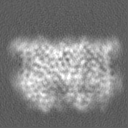
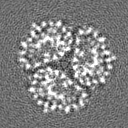
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : Trimeric maltoporin (LamB protein)
| Entire | Name: Trimeric maltoporin (LamB protein) |
|---|---|
| Components |
|
-Supramolecule #1: Trimeric maltoporin (LamB protein)
| Supramolecule | Name: Trimeric maltoporin (LamB protein) / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Escherichia coli (E. coli) |
-Macromolecule #1: Maltoporin
| Macromolecule | Name: Maltoporin / type: protein_or_peptide / ID: 1 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Escherichia coli (E. coli) |
| Molecular weight | Theoretical: 47.425785 KDa |
| Sequence | String: VDFHGYARSG IGWTGSGGEQ QCFQTTGAQS KYRLGNECET YAELKLGQEV WKEGDKSFYF DTNVAYSVAQ QNDWEATDPA FREANVQGK NLIEWLPGST IWAGKRFYQR HDVHMIDFYY WDISGPGAGL ENIDVGFGKL SLAATRSSEA GGSSSFASNN I YDYTNETA ...String: VDFHGYARSG IGWTGSGGEQ QCFQTTGAQS KYRLGNECET YAELKLGQEV WKEGDKSFYF DTNVAYSVAQ QNDWEATDPA FREANVQGK NLIEWLPGST IWAGKRFYQR HDVHMIDFYY WDISGPGAGL ENIDVGFGKL SLAATRSSEA GGSSSFASNN I YDYTNETA NDVFDVRLAQ MEINPGGTLE LGVDYGRANL RDNYRLVDGA SKDGWLFTAE HTQSVLKGFN KFVVQYATDS MT SQGKGLS QGSGVAFDNE KFAYNINNNG HMLRILDHGA ISMGDNWDMM YVGMYQDINW DNDNGTKWWT VGIRPMYKWT PIM STVMEI GYDNVESQRT GDKNNQYKIT LAQQWQAGDS IWSRPAIRVF ATYAKWDEKW GYDYTGNADN NANFGKAVPA DFNG GSFGR GDSDEWTFGA QMEIWW UniProtKB: Maltoporin |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3 mg/mL |
|---|---|
| Buffer | pH: 7.4 |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Nominal defocus max: 2.8000000000000003 µm / Nominal defocus min: 1.4000000000000001 µm |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Average electron dose: 42.0 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Startup model | Type of model: PDB ENTRY PDB model - PDB ID: |
|---|---|
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD |
| Final reconstruction | Resolution.type: BY AUTHOR / Resolution: 3.0 Å / Resolution method: FSC 0.143 CUT-OFF / Number images used: 579000 |
| FSC plot (resolution estimation) | 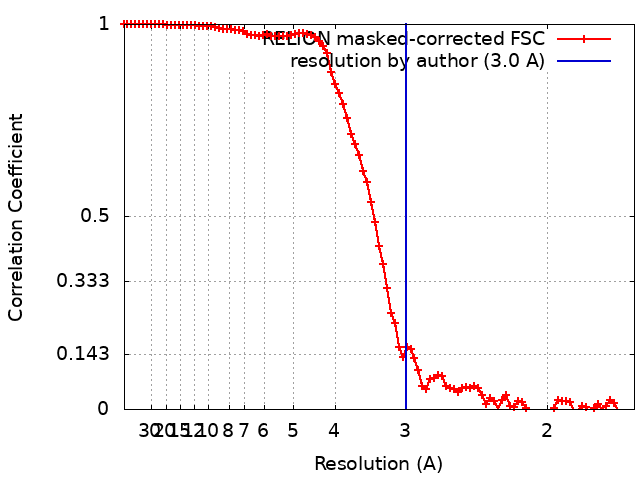 |

