 ムービー
ムービー コントローラー
コントローラー

[日本語] English
 万見
万見- EMDB-13634: Structure of hexameric S-layer protein from Haloferax volcanii archaea -
+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-13634 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Structure of hexameric S-layer protein from Haloferax volcanii archaea | ||||||||||||
 マップデータ マップデータ | Full map, non-sharpened, non post-processed | ||||||||||||
 試料 試料 |
| ||||||||||||
| 機能・相同性 | Surface glycoprotein signal peptide / Major cell surface glycoprotein / PGF-CTERM archaeal protein-sorting signal / PGF-CTERM motif /  S層 / cell wall organization / extracellular region / 細胞膜 / Cell surface glycoprotein S層 / cell wall organization / extracellular region / 細胞膜 / Cell surface glycoprotein 機能・相同性情報 機能・相同性情報 | ||||||||||||
| 生物種 |  Haloferax volcanii DS2 (古細菌) Haloferax volcanii DS2 (古細菌) | ||||||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.46 Å | ||||||||||||
 データ登録者 データ登録者 | von Kuegelgen A / Bharat TAM | ||||||||||||
| 資金援助 |  英国, 3件 英国, 3件
| ||||||||||||
 引用 引用 | ジャーナル: Cell Rep / 年: 2021 タイトル: Complete atomic structure of a native archaeal cell surface. 著者: Andriko von Kügelgen / Vikram Alva / Tanmay A M Bharat /  要旨: Many prokaryotic cells are covered by an ordered, proteinaceous, sheet-like structure called a surface layer (S-layer). S-layer proteins (SLPs) are usually the highest copy number macromolecules in ...Many prokaryotic cells are covered by an ordered, proteinaceous, sheet-like structure called a surface layer (S-layer). S-layer proteins (SLPs) are usually the highest copy number macromolecules in prokaryotes, playing critical roles in cellular physiology such as blocking predators, scaffolding membranes, and facilitating environmental interactions. Using electron cryomicroscopy of two-dimensional sheets, we report the atomic structure of the S-layer from the archaeal model organism Haloferax volcanii. This S-layer consists of a hexagonal array of tightly interacting immunoglobulin-like domains, which are also found in SLPs across several classes of archaea. Cellular tomography reveal that the S-layer is nearly continuous on the cell surface, completed by pentameric defects in the hexagonal lattice. We further report the atomic structure of the SLP pentamer, which shows markedly different relative arrangements of SLP domains needed to complete the S-layer. Our structural data provide a framework for understanding cell surfaces of archaea at the atomic level. | ||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_13634.map.gz | 188.8 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-13634-v30.xmlemd-13634.xml | 29.3 KB 29.3 KB | 表示 表示 | EMDBヘッダ |
| 画像 | 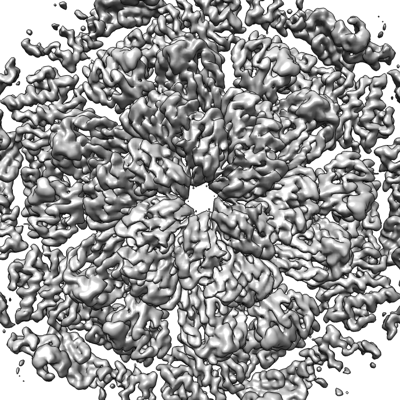 emd_13634.png emd_13634.png | 235.4 KB | ||
| マスクデータ | emd_13634_msk_1.map | 244.1 MB | マスクマップ | |
| その他 | emd_13634_additional_1.map.gzemd_13634_half_map_1.map.gzemd_13634_half_map_2.map.gz | 212 MB 191.3 MB 191.3 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-13634ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13634 http://ftp.pdbj.org/pub/emdb/structures/EMD-13634ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13634 | HTTPS FTP |
-関連構造データ












-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_13634.map.gz / 形式: CCP4 / 大きさ: 244.1 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Full map, non-sharpened, non post-processed | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-添付データ
-マスク #1
| ファイル | emd_13634_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 |
| ||||||||||||
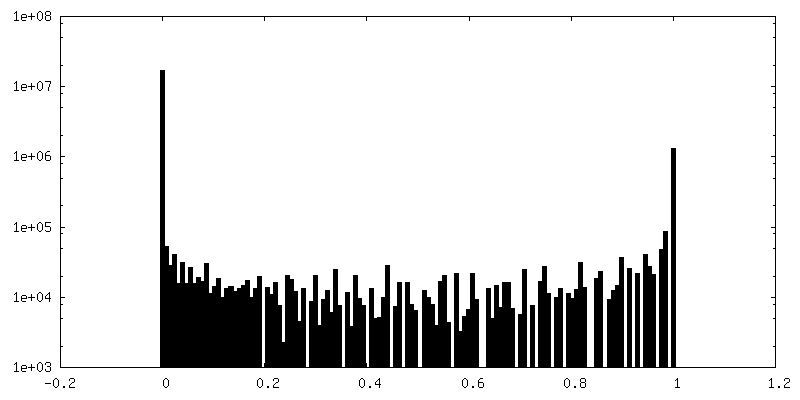
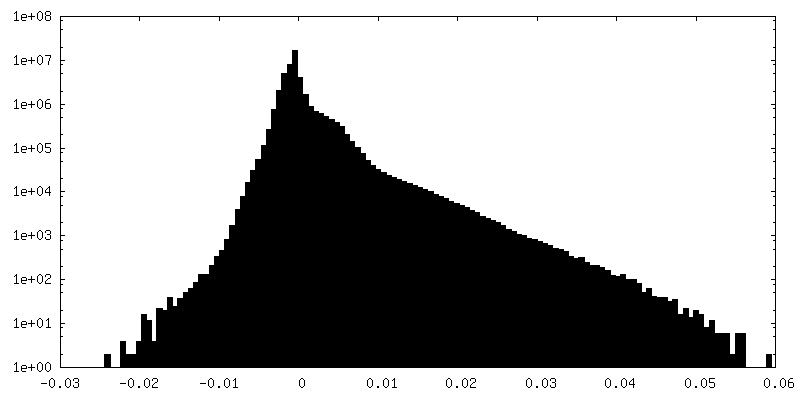
| 密度ヒストグラム |
 Z
Z Y
Y X
X








-追加マップ: Density modified map using deepEMhancer (Sanchez-Garcia et al 2021)
| ファイル | emd_13634_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Density modified map using deepEMhancer (Sanchez-Garcia et al 2021) | ||||||||||||
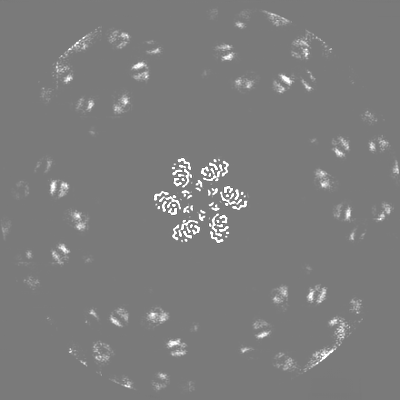
| 投影像・断面図 |
| ||||||||||||
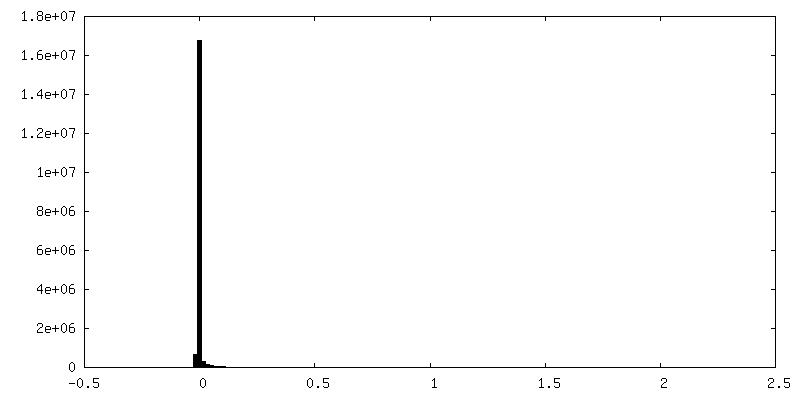
| 密度ヒストグラム |






-ハーフマップ: Half map1, non-sharpened, non post-processed
| ファイル | emd_13634_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half map1, non-sharpened, non post-processed | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |







-ハーフマップ: Half map2, non-sharpened, non post-processed
| ファイル | emd_13634_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half map2, non-sharpened, non post-processed | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |






- 試料の構成要素
試料の構成要素
-全体 : Structure of hexameric S-layer protein csg
| 全体 | 名称: Structure of hexameric S-layer protein csg |
|---|---|
| 要素 |
|
-超分子 #1: Structure of hexameric S-layer protein csg
| 超分子 | 名称: Structure of hexameric S-layer protein csg / タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: #1 / 詳細: Structure of hexameric S-layer protein csg |
|---|---|
| 由来(天然) | 生物種: Haloferax volcanii DS2 (古細菌) / 細胞中の位置: Cell surface |
-分子 #1: Cell surface glycoprotein
| 分子 | 名称: Cell surface glycoprotein / タイプ: protein_or_peptide / ID: 1 / コピー数: 6 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: Haloferax volcanii DS2 (古細菌) |
| 分子量 | 理論値: 81.755602 KDa |
| 配列 | 文字列: ERGNLDADSE SFNKTIQSGD RVFLGEEIST DAGLGASNPL LTGTAGNSEG VSLDLSSPIP QTTENQPLGT YDVDGSGSAT TPNVTLLAP RITDSEILTS SGGDVTGSAI SSSDAGNLYV NADYNYESAE KVEVTVEDPS GTDITNEVLS GTDTFVDDGS I GSTSSTGG ...文字列: ERGNLDADSE SFNKTIQSGD RVFLGEEIST DAGLGASNPL LTGTAGNSEG VSLDLSSPIP QTTENQPLGT YDVDGSGSAT TPNVTLLAP RITDSEILTS SGGDVTGSAI SSSDAGNLYV NADYNYESAE KVEVTVEDPS GTDITNEVLS GTDTFVDDGS I GSTSSTGG GVGIDMSDQD AGEYTIILEG AEDLDFGDAT ETMTLTISSQ DEIGIELDSE SVTQGTDVQY TVTNGIDGNE HV VAMDLSD LQNDATTEQA KEVFRNIGDT SEVGIANSSA TNTSGSSTGP TVETADIAYA VVEIDGASAV GGIETQYLDD SEV DLEVYD AGVSATAAVG QDATNDITLT IEEGGTTLSS PTGQYVVGSE VDINGTATSS DSVAIYVRDD GDWQLLEIGG DNEI SVDSD DTFEEEDIAL SGLSGDGSSI LSLTGTYRIG VIDASDADVG GDGSVDDSLT TSEFTSGVSS SNSIRVTDQA LTGQF TTIN GQVAPVETGT VDINGTASGA NSVLVIFVDE RGNVNYQEVS VDSDGTYDED DITVGLTQGR VTAHILSVGR DSAIGD GSL PSGPSNGATL NDLTGYLDTL DQNNNNGEQI NELIASETVD ETASDDLIVT ETFRLAESST SIDSIYPDAA EAAGINP VA TGETMVIAGS TNLKPDDNTI SIEVTNEDGT SVALEDTDEW NNDGQWMVEI DTTDFETGTF TVEADDGDNT DTVNVEVV S EREDTTTSSD NATDTTTTTD GPTETTTTAE PTETTEEPTE ETTTSSNTPG FGIAVALVAL VGAALLALRR EN |
-分子 #2: CALCIUM ION
| 分子 | 名称: CALCIUM ION / タイプ: ligand / ID: 2 / コピー数: 18 / 式: CA |
|---|---|
| 分子量 | 理論値: 40.078 Da |
-分子 #3: beta-D-glucopyranose
| 分子 | 名称: beta-D-glucopyranose / タイプ: ligand / ID: 3 / コピー数: 18 / 式: BGC |
|---|---|
| 分子量 | 理論値: 180.156 Da |
| Chemical component information |  ChemComp-BGC: |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | 2D array |
-試料調製
| 濃度 | 3.2 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 緩衝液 | pH: 7.5 構成要素:
詳細: Buffer solutions were prepared fresh from sterile filtered concentrated stocksolutions. Solutions were filtered through a 0.22 um filter to avoid microbial contamination and degassed using a ...詳細: Buffer solutions were prepared fresh from sterile filtered concentrated stocksolutions. Solutions were filtered through a 0.22 um filter to avoid microbial contamination and degassed using a vacuum fold pump. The pH of the HEPES stock solution was adjusted with sodium hydroxide at 4 deg C. 15 mM Calcium chloride was added 15 minutes before vitrification. | |||||||||||||||
| グリッド | モデル: Quantifoil R2/2 / 材質: COPPER/RHODIUM / メッシュ: 200 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: HOLEY / 前処理 - タイプ: GLOW DISCHARGE / 前処理 - 雰囲気: AIR / 詳細: 20 seconds, 15 mA | |||||||||||||||
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 283.15 K / 装置: FEI VITROBOT MARK IV 詳細: Vitrobot options: Blot time 4.5 seconds, Blot force -10,1, Wait time 10 seconds, Drain time 0.5 seconds. | |||||||||||||||
| 詳細 | Purified csg protein mixed with 15 mM CaCl2 after 15 minutes incubation. |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 電子線 | 加速電圧: 300 kV / 電子線源: FIELD EMISSION GUN |
| 電子光学系 | C2レンズ絞り径: 50.0 µm / 最大 デフォーカス(補正後): 4.0 µm / 最小 デフォーカス(補正後): 1.0 µm / 倍率(補正後): 81000 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / 最大 デフォーカス(公称値): 4.0 µm / 最小 デフォーカス(公称値): 1.0 µm / 倍率(公称値): 81000 |
| 特殊光学系 | エネルギーフィルター - 名称: GIF Quantum LS / エネルギーフィルター - スリット幅: 20 eV |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 温度 | 最低: 70.0 K / 最高: 70.0 K |
| 詳細 | EPU software with faster acquisition mode AFIS (Aberration Free Image Shift). |
| 撮影 | フィルム・検出器のモデル: GATAN K3 BIOQUANTUM (6k x 4k) デジタル化 - サイズ - 横: 5760 pixel / デジタル化 - サイズ - 縦: 4092 pixel / 撮影したグリッド数: 2 / 実像数: 18468 / 平均露光時間: 3.4 sec. / 平均電子線量: 51.441 e/Å2 詳細: Images were collected in two sessions movie-mode and subjected to 3.4 seconds of exposure where a total dose of 49 or 51.441 e-/A2 was applied, and 40 frames were recorded per movie. A total ...詳細: Images were collected in two sessions movie-mode and subjected to 3.4 seconds of exposure where a total dose of 49 or 51.441 e-/A2 was applied, and 40 frames were recorded per movie. A total of 18468 movies were collected in two sessions with the same microscope and settings. |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| 粒子像選択 | 選択した数: 10558369 詳細: Top and tilted views were manually picked at the central hexameric axis. Manually picked particles were extracted in 4x downsampled 100 x 100 boxes and classified using reference-free 2D ...詳細: Top and tilted views were manually picked at the central hexameric axis. Manually picked particles were extracted in 4x downsampled 100 x 100 boxes and classified using reference-free 2D classification inside RELION3.1 (Zivanov et al., 2020). Class averages centered at a hexameric axis were used to automatically pick particles inside RELION3.1. Automatically picked particles were extracted in 4x downsampled 100x100 pixel boxes and classified using reference-free 2D classification. Particle coordinates belonging to class averages centered at the hexameric axis were used to train TOPAZ (Bepler et al., 2019) in 5x downsampled micrographs with the neural network architecture ResNet8. For the final reconstruction, particles were picked using TOPAZ and the previously trained neural network above. Additionally, top and bottom views were picked using the reference-based autopicker inside RELION3.1, which were not readily identified by TOPAZ. Particles were extracted in 4x downsampled 100 x 100 boxes and classified using reference-free 2D classification inside RELION3.1. Particles belonging to class averages centered at the hexameric axis were combined, and particles within 100 angstrom were removed to prevent duplication after alignment. |
|---|---|
| CTF補正 | ソフトウェア - 名称: CTFFIND (ver. 4.1.13) ソフトウェア - 詳細: CTFFIND4 was used as implemented in RELION 3.1 詳細: RELION refinement with in-built CTF correction. The function is similar to a Wiener filter, so amplitude correction included. |
| 初期モデル | モデルのタイプ: OTHER 詳細: Initial model generation from cryo-ET data was performed using the RELION sub-tomogram averaging pipeline (Bharat et al., 2015; Bharat and Scheres, 2016) from subtomogram averaging of the inverted S-layer tubes. |
| 初期 角度割当 | タイプ: MAXIMUM LIKELIHOOD / ソフトウェア - 名称: RELION (ver. 3.1) / 詳細: Angle assignment was performed within RELION3.1 |
| 最終 3次元分類 | クラス数: 6 / ソフトウェア - 名称: RELION (ver. 3.1) 詳細: All resulting particles, side views from TOPAZ picking and top/bottom views from RELION picking, were then re-extracted in 4x downsampled 100 x 100 boxes and were subjected to 3D ...詳細: All resulting particles, side views from TOPAZ picking and top/bottom views from RELION picking, were then re-extracted in 4x downsampled 100 x 100 boxes and were subjected to 3D classification using a 60 angstrom lowpass filtered reference map from subtomogram averaging of the inverted S-layer tubes. Particles from classes with the same curvature were combined and re-extracted in 400 x 400 boxes. |
| 最終 角度割当 | タイプ: MAXIMUM LIKELIHOOD / ソフトウェア - 名称: RELION (ver. 3.1) / 詳細: Angle assignment was performed within RELION3.1 |
| 最終 再構成 | 使用したクラス数: 1 / 想定した対称性 - 点群: C6 (6回回転対称) / アルゴリズム: FOURIER SPACE / 解像度のタイプ: BY AUTHOR / 解像度: 3.46 Å / 解像度の算出法: FSC 0.143 CUT-OFF / ソフトウェア - 名称: RELION (ver. 3.1) 詳細: Particles from classes with the same curvature were combined, re-extracted in 400 x 400 boxes and subjected to a focused 3D auto refinement on the central 6 subunits using the scaled and ...詳細: Particles from classes with the same curvature were combined, re-extracted in 400 x 400 boxes and subjected to a focused 3D auto refinement on the central 6 subunits using the scaled and lowpass filtered output from the 3D classification as a starting model. Per-particle defocus, anisotropy magnification and higher-order aberrations were refined inside RELION3.1, followed by another round of focused 3D auto refinement and Bayesian particle polishing (Zivanov et al., 2020). 使用した粒子像数: 1087798 |
| 詳細 | Movies were clustered into optics groups based on the XML meta-data of the data-collection software EPU (ThermoFisher) using a k-means algorithm implemented in EPU_group_AFIS (https://github.com/DustinMorado/EPU_group_AFIS). Imported movies were motion-corrected, dose weighted, and Fourier cropped (2x) with MotionCor2 (Zheng et al., 2017) implemented in RELION3.1 (Zivanov et al., 2018). Contrast transfer functions (CTFs) of the resulting motion-corrected micrographs were estimated using CTFFIND4 (Rohou and Grigorieff, 2015). |
-原子モデル構築 1
| 詳細 | The boundaries of the six Ig-like domains, D1-D6, were predicted using HHpred (Steinegger et al., 2019) in default settings within the MPI Bioinformatics Toolkit (Zimmermann et al., 2018). Subsequently, structural models for these domains were built using the Robetta structure prediction server, employing the deep learning-based modelling method TrRosetta (Yang et al., 2020). The obtained structural models of domains D3-D6 resulted in an overall fit into the hexameric cryo-EM map of csg from the reconstituted sheets. D1-D2 deviated significantly from any obtained homology models, and for those domains, the carbon backbone of the csg protein was manually traced through a single subunit of the hexameric cryo-EM density using Coot (Emsley and Cowtan, 2004). Due to the edge effect of the box used in the refinement of the 3.5 angstrom map, parts of D6 displayed edge artefacts. These artefacts were removed using single-particle cryo-EM refinement in a larger box, which led to an overall slightly lower resolution (3.8 angstrom) but allowed fitting of the D6 homology model unambiguously. Following initial manual building (for D1-D2) or fitting in of structural models (for D3-D6), side chains were assigned in regions with density corresponding to characteristic aromatic residues allowing us to deduce the register of the amino acid sequence in the map. Another important check of the model building was the position of known glycan positions, which were readily assigned based on large unexplained densities on characteristic asparagine residues. The atomic model was then placed into the hexameric map in six copies and subjected to several rounds of refinement using refmac5 (Murshudov et al., 2011) inside the CCP-EM software suite (Burnley et al., 2017) and PHENIX (Liebschner et al., 2019), followed by manually rebuilding in Coot (Emsley and Cowtan, 2004). Model validation was performed in PHENIX and CCP-EM. |
|---|---|
| 精密化 | 空間: REAL / プロトコル: AB INITIO MODEL / 温度因子: 143.26 / 当てはまり具合の基準: Best Fit |
| 得られたモデル |  PDB-7ptr: |
