 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-13634: Structure of hexameric S-layer protein from Haloferax volcanii archaea -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-13634 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of hexameric S-layer protein from Haloferax volcanii archaea | ||||||||||||
 Map data Map data | Full map, non-sharpened, non post-processed | ||||||||||||
 Sample Sample |
| ||||||||||||
| Function / homology | Surface glycoprotein signal peptide / Major cell surface glycoprotein / PGF-CTERM archaeal protein-sorting signal / PGF-CTERM motif /  S-layer / cell wall organization / extracellular region / plasma membrane / Cell surface glycoprotein S-layer / cell wall organization / extracellular region / plasma membrane / Cell surface glycoprotein Function and homology information Function and homology information | ||||||||||||
| Biological species |  Haloferax volcanii DS2 (archaea) Haloferax volcanii DS2 (archaea) | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.46 Å | ||||||||||||
 Authors Authors | von Kuegelgen A / Bharat TAM | ||||||||||||
| Funding support |  United Kingdom, 3 items United Kingdom, 3 items
| ||||||||||||
 Citation Citation | Journal: Cell Rep / Year: 2021 Title: Complete atomic structure of a native archaeal cell surface. Authors: Andriko von Kügelgen / Vikram Alva / Tanmay A M Bharat /  Abstract: Many prokaryotic cells are covered by an ordered, proteinaceous, sheet-like structure called a surface layer (S-layer). S-layer proteins (SLPs) are usually the highest copy number macromolecules in ...Many prokaryotic cells are covered by an ordered, proteinaceous, sheet-like structure called a surface layer (S-layer). S-layer proteins (SLPs) are usually the highest copy number macromolecules in prokaryotes, playing critical roles in cellular physiology such as blocking predators, scaffolding membranes, and facilitating environmental interactions. Using electron cryomicroscopy of two-dimensional sheets, we report the atomic structure of the S-layer from the archaeal model organism Haloferax volcanii. This S-layer consists of a hexagonal array of tightly interacting immunoglobulin-like domains, which are also found in SLPs across several classes of archaea. Cellular tomography reveal that the S-layer is nearly continuous on the cell surface, completed by pentameric defects in the hexagonal lattice. We further report the atomic structure of the SLP pentamer, which shows markedly different relative arrangements of SLP domains needed to complete the S-layer. Our structural data provide a framework for understanding cell surfaces of archaea at the atomic level. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_13634.map.gz | 188.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-13634-v30.xmlemd-13634.xml | 29.3 KB 29.3 KB | Display Display | EMDB header |
| Images | 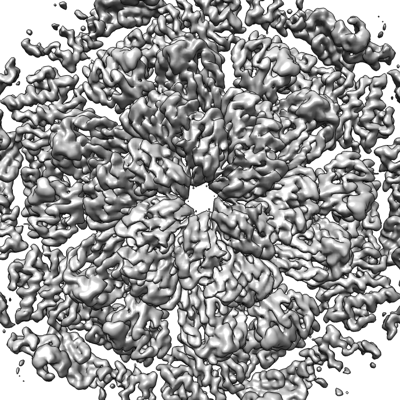 emd_13634.png emd_13634.png | 235.4 KB | ||
| Masks | emd_13634_msk_1.map | 244.1 MB | Mask map | |
| Others | emd_13634_additional_1.map.gzemd_13634_half_map_1.map.gzemd_13634_half_map_2.map.gz | 212 MB 191.3 MB 191.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-13634ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13634 http://ftp.pdbj.org/pub/emdb/structures/EMD-13634ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13634 | HTTPS FTP |
-Related structure data
| Related structure data |  7ptrMC  7ptpC  7pttC  7ptuC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_13634.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Full map, non-sharpened, non post-processed | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
-Mask #1
| File | emd_13634_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
 Z
Z Y
Y X
X








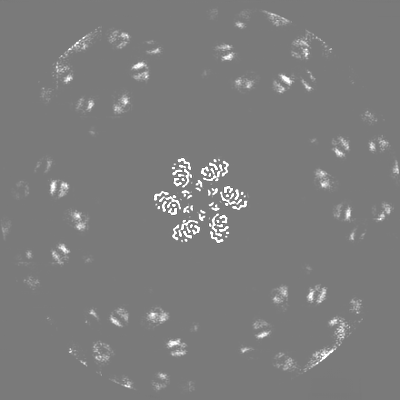
-Additional map: Density modified map using deepEMhancer (Sanchez-Garcia et al 2021)
| File | emd_13634_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Density modified map using deepEMhancer (Sanchez-Garcia et al 2021) | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: Half map1, non-sharpened, non post-processed
| File | emd_13634_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map1, non-sharpened, non post-processed | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Half map: Half map2, non-sharpened, non post-processed
| File | emd_13634_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map2, non-sharpened, non post-processed | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : Structure of hexameric S-layer protein csg
| Entire | Name: Structure of hexameric S-layer protein csg |
|---|---|
| Components |
|
-Supramolecule #1: Structure of hexameric S-layer protein csg
| Supramolecule | Name: Structure of hexameric S-layer protein csg / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 / Details: Structure of hexameric S-layer protein csg |
|---|---|
| Source (natural) | Organism: Haloferax volcanii DS2 (archaea) / Location in cell: Cell surface |
-Macromolecule #1: Cell surface glycoprotein
| Macromolecule | Name: Cell surface glycoprotein / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Haloferax volcanii DS2 (archaea) |
| Molecular weight | Theoretical: 81.755602 KDa |
| Sequence | String: ERGNLDADSE SFNKTIQSGD RVFLGEEIST DAGLGASNPL LTGTAGNSEG VSLDLSSPIP QTTENQPLGT YDVDGSGSAT TPNVTLLAP RITDSEILTS SGGDVTGSAI SSSDAGNLYV NADYNYESAE KVEVTVEDPS GTDITNEVLS GTDTFVDDGS I GSTSSTGG ...String: ERGNLDADSE SFNKTIQSGD RVFLGEEIST DAGLGASNPL LTGTAGNSEG VSLDLSSPIP QTTENQPLGT YDVDGSGSAT TPNVTLLAP RITDSEILTS SGGDVTGSAI SSSDAGNLYV NADYNYESAE KVEVTVEDPS GTDITNEVLS GTDTFVDDGS I GSTSSTGG GVGIDMSDQD AGEYTIILEG AEDLDFGDAT ETMTLTISSQ DEIGIELDSE SVTQGTDVQY TVTNGIDGNE HV VAMDLSD LQNDATTEQA KEVFRNIGDT SEVGIANSSA TNTSGSSTGP TVETADIAYA VVEIDGASAV GGIETQYLDD SEV DLEVYD AGVSATAAVG QDATNDITLT IEEGGTTLSS PTGQYVVGSE VDINGTATSS DSVAIYVRDD GDWQLLEIGG DNEI SVDSD DTFEEEDIAL SGLSGDGSSI LSLTGTYRIG VIDASDADVG GDGSVDDSLT TSEFTSGVSS SNSIRVTDQA LTGQF TTIN GQVAPVETGT VDINGTASGA NSVLVIFVDE RGNVNYQEVS VDSDGTYDED DITVGLTQGR VTAHILSVGR DSAIGD GSL PSGPSNGATL NDLTGYLDTL DQNNNNGEQI NELIASETVD ETASDDLIVT ETFRLAESST SIDSIYPDAA EAAGINP VA TGETMVIAGS TNLKPDDNTI SIEVTNEDGT SVALEDTDEW NNDGQWMVEI DTTDFETGTF TVEADDGDNT DTVNVEVV S EREDTTTSSD NATDTTTTTD GPTETTTTAE PTETTEEPTE ETTTSSNTPG FGIAVALVAL VGAALLALRR EN |
-Macromolecule #2: CALCIUM ION
| Macromolecule | Name: CALCIUM ION / type: ligand / ID: 2 / Number of copies: 18 / Formula: CA |
|---|---|
| Molecular weight | Theoretical: 40.078 Da |
-Macromolecule #3: beta-D-glucopyranose
| Macromolecule | Name: beta-D-glucopyranose / type: ligand / ID: 3 / Number of copies: 18 / Formula: BGC |
|---|---|
| Molecular weight | Theoretical: 180.156 Da |
| Chemical component information | 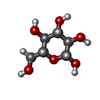 ChemComp-BGC: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | 2D array |
-Sample preparation
| Concentration | 3.2 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
Details: Buffer solutions were prepared fresh from sterile filtered concentrated stocksolutions. Solutions were filtered through a 0.22 um filter to avoid microbial contamination and degassed using a ...Details: Buffer solutions were prepared fresh from sterile filtered concentrated stocksolutions. Solutions were filtered through a 0.22 um filter to avoid microbial contamination and degassed using a vacuum fold pump. The pH of the HEPES stock solution was adjusted with sodium hydroxide at 4 deg C. 15 mM Calcium chloride was added 15 minutes before vitrification. | |||||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER/RHODIUM / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR / Details: 20 seconds, 15 mA | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 283.15 K / Instrument: FEI VITROBOT MARK IV Details: Vitrobot options: Blot time 4.5 seconds, Blot force -10,1, Wait time 10 seconds, Drain time 0.5 seconds. | |||||||||||||||
| Details | Purified csg protein mixed with 15 mM CaCl2 after 15 minutes incubation. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Calibrated defocus max: 4.0 µm / Calibrated defocus min: 1.0 µm / Calibrated magnification: 81000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 4.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 81000 |
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Slit width: 20 eV |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Temperature | Min: 70.0 K / Max: 70.0 K |
| Details | EPU software with faster acquisition mode AFIS (Aberration Free Image Shift). |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 2 / Number real images: 18468 / Average exposure time: 3.4 sec. / Average electron dose: 51.441 e/Å2 Details: Images were collected in two sessions movie-mode and subjected to 3.4 seconds of exposure where a total dose of 49 or 51.441 e-/A2 was applied, and 40 frames were recorded per movie. A total ...Details: Images were collected in two sessions movie-mode and subjected to 3.4 seconds of exposure where a total dose of 49 or 51.441 e-/A2 was applied, and 40 frames were recorded per movie. A total of 18468 movies were collected in two sessions with the same microscope and settings. |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 10558369 Details: Top and tilted views were manually picked at the central hexameric axis. Manually picked particles were extracted in 4x downsampled 100 x 100 boxes and classified using reference-free 2D ...Details: Top and tilted views were manually picked at the central hexameric axis. Manually picked particles were extracted in 4x downsampled 100 x 100 boxes and classified using reference-free 2D classification inside RELION3.1 (Zivanov et al., 2020). Class averages centered at a hexameric axis were used to automatically pick particles inside RELION3.1. Automatically picked particles were extracted in 4x downsampled 100x100 pixel boxes and classified using reference-free 2D classification. Particle coordinates belonging to class averages centered at the hexameric axis were used to train TOPAZ (Bepler et al., 2019) in 5x downsampled micrographs with the neural network architecture ResNet8. For the final reconstruction, particles were picked using TOPAZ and the previously trained neural network above. Additionally, top and bottom views were picked using the reference-based autopicker inside RELION3.1, which were not readily identified by TOPAZ. Particles were extracted in 4x downsampled 100 x 100 boxes and classified using reference-free 2D classification inside RELION3.1. Particles belonging to class averages centered at the hexameric axis were combined, and particles within 100 angstrom were removed to prevent duplication after alignment. |
|---|---|
| CTF correction | Software - Name: CTFFIND (ver. 4.1.13) Software - details: CTFFIND4 was used as implemented in RELION 3.1 Details: RELION refinement with in-built CTF correction. The function is similar to a Wiener filter, so amplitude correction included. |
| Startup model | Type of model: OTHER Details: Initial model generation from cryo-ET data was performed using the RELION sub-tomogram averaging pipeline (Bharat et al., 2015; Bharat and Scheres, 2016) from subtomogram averaging of the inverted S-layer tubes. |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) / Details: Angle assignment was performed within RELION3.1 |
| Final 3D classification | Number classes: 6 / Software - Name: RELION (ver. 3.1) Details: All resulting particles, side views from TOPAZ picking and top/bottom views from RELION picking, were then re-extracted in 4x downsampled 100 x 100 boxes and were subjected to 3D ...Details: All resulting particles, side views from TOPAZ picking and top/bottom views from RELION picking, were then re-extracted in 4x downsampled 100 x 100 boxes and were subjected to 3D classification using a 60 angstrom lowpass filtered reference map from subtomogram averaging of the inverted S-layer tubes. Particles from classes with the same curvature were combined and re-extracted in 400 x 400 boxes. |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) / Details: Angle assignment was performed within RELION3.1 |
| Final reconstruction | Number classes used: 1 / Applied symmetry - Point group: C6 (6 fold cyclic) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 3.46 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 3.1) Details: Particles from classes with the same curvature were combined, re-extracted in 400 x 400 boxes and subjected to a focused 3D auto refinement on the central 6 subunits using the scaled and ...Details: Particles from classes with the same curvature were combined, re-extracted in 400 x 400 boxes and subjected to a focused 3D auto refinement on the central 6 subunits using the scaled and lowpass filtered output from the 3D classification as a starting model. Per-particle defocus, anisotropy magnification and higher-order aberrations were refined inside RELION3.1, followed by another round of focused 3D auto refinement and Bayesian particle polishing (Zivanov et al., 2020). Number images used: 1087798 |
| Details | Movies were clustered into optics groups based on the XML meta-data of the data-collection software EPU (ThermoFisher) using a k-means algorithm implemented in EPU_group_AFIS (https://github.com/DustinMorado/EPU_group_AFIS). Imported movies were motion-corrected, dose weighted, and Fourier cropped (2x) with MotionCor2 (Zheng et al., 2017) implemented in RELION3.1 (Zivanov et al., 2018). Contrast transfer functions (CTFs) of the resulting motion-corrected micrographs were estimated using CTFFIND4 (Rohou and Grigorieff, 2015). |
-Atomic model buiding 1
| Details | The boundaries of the six Ig-like domains, D1-D6, were predicted using HHpred (Steinegger et al., 2019) in default settings within the MPI Bioinformatics Toolkit (Zimmermann et al., 2018). Subsequently, structural models for these domains were built using the Robetta structure prediction server, employing the deep learning-based modelling method TrRosetta (Yang et al., 2020). The obtained structural models of domains D3-D6 resulted in an overall fit into the hexameric cryo-EM map of csg from the reconstituted sheets. D1-D2 deviated significantly from any obtained homology models, and for those domains, the carbon backbone of the csg protein was manually traced through a single subunit of the hexameric cryo-EM density using Coot (Emsley and Cowtan, 2004). Due to the edge effect of the box used in the refinement of the 3.5 angstrom map, parts of D6 displayed edge artefacts. These artefacts were removed using single-particle cryo-EM refinement in a larger box, which led to an overall slightly lower resolution (3.8 angstrom) but allowed fitting of the D6 homology model unambiguously. Following initial manual building (for D1-D2) or fitting in of structural models (for D3-D6), side chains were assigned in regions with density corresponding to characteristic aromatic residues allowing us to deduce the register of the amino acid sequence in the map. Another important check of the model building was the position of known glycan positions, which were readily assigned based on large unexplained densities on characteristic asparagine residues. The atomic model was then placed into the hexameric map in six copies and subjected to several rounds of refinement using refmac5 (Murshudov et al., 2011) inside the CCP-EM software suite (Burnley et al., 2017) and PHENIX (Liebschner et al., 2019), followed by manually rebuilding in Coot (Emsley and Cowtan, 2004). Model validation was performed in PHENIX and CCP-EM. |
|---|---|
| Refinement | Space: REAL / Protocol: AB INITIO MODEL / Overall B value: 143.26 / Target criteria: Best Fit |
| Output model | PDB-7ptr: |
