 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3zuh: Negative stain EM Map of the AAA protein CbbX, a red-type Rubisco... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3zuh | ||||||
|---|---|---|---|---|---|---|---|
| Title | Negative stain EM Map of the AAA protein CbbX, a red-type Rubisco activase from R. sphaeroides | ||||||
 Components Components | PROTEIN CBBX | ||||||
 Keywords Keywords | ATP BINDING PROTEIN /  AAA+ PROTEIN AAA+ PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  RHODOBACTER SPHAEROIDES (bacteria) RHODOBACTER SPHAEROIDES (bacteria) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / negative staining / Resolution: 21 Å | ||||||
 Authors Authors | Mueller-Cajar, O. / Stotz, M. / Wendler, P. / Hartl, F.U. / Bracher, A. / Hayer-Hartl, M. | ||||||
 Citation Citation | Journal: Nature / Year: 2011 Title: Structure and function of the AAA+ protein CbbX, a red-type Rubisco activase. Authors: Oliver Mueller-Cajar / Mathias Stotz / Petra Wendler / F Ulrich Hartl / Andreas Bracher / Manajit Hayer-Hartl /  Abstract: Ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco) catalyses the fixation of atmospheric CO(2) in photosynthesis, but tends to form inactive complexes with its substrate ribulose 1,5- ...Ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco) catalyses the fixation of atmospheric CO(2) in photosynthesis, but tends to form inactive complexes with its substrate ribulose 1,5-bisphosphate (RuBP). In plants, Rubisco is reactivated by the AAA(+) (ATPases associated with various cellular activities) protein Rubisco activase (Rca), but no such protein is known for the Rubisco of red algae. Here we identify the protein CbbX as an activase of red-type Rubisco. The 3.0-Å crystal structure of unassembled CbbX from Rhodobacter sphaeroides revealed an AAA(+) protein architecture. Electron microscopy and biochemical analysis showed that ATP and RuBP must bind to convert CbbX into functionally active, hexameric rings. The CbbX ATPase is strongly stimulated by RuBP and Rubisco. Mutational analysis suggests that CbbX functions by transiently pulling the carboxy-terminal peptide of the Rubisco large subunit into the hexamer pore, resulting in the release of the inhibitory RuBP. Understanding Rubisco activation may facilitate efforts to improve CO(2) uptake and biomass production by photosynthetic organisms. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3zuh.cif.gz | 298.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3zuh.ent.gz | 252.6 KB | Display | PDB format |
| PDBx/mmJSON format | 3zuh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zu/3zuhftp://data.pdbj.org/pub/pdb/validation_reports/zu/3zuh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1932MC  3sykC  3sylC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 32612.963 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Details: THE PROTEIN IS BOUND TO RIBULOSE-1,5-BISPHOSPHATE / Source: (gene. exp.) RHODOBACTER SPHAEROIDES (bacteria) / Production host: ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P95648#2: Sugar | ChemComp-RUB / Ribulose 1,5-bisphosphate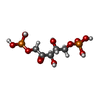  Type: saccharideCarbohydrate / Mass: 310.090 Da / Num. of mol.: 6 Type: saccharideCarbohydrate / Mass: 310.090 Da / Num. of mol.: 6Source method: isolated from a genetically manipulated source Formula: C5H12O11P2 #3: Chemical | ChemComp-ADP / Adenosine diphosphate  Mass: 427.201 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YMNonpolymer details | ADENOSINE-5'-DIPHOSPHAT | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: PROTEIN CBBX / Type: COMPLEX |
|---|---|
| Buffer solution | Name: 20 MM TRIS PH 8.0, 50 MM NACL, 5MM MGCL2, 1MM RIBULOSE-1,5- BISPHOSPHATE, 1MM ATP/ATPGAMMAS pH: 8 Details: 20 MM TRIS PH 8.0, 50 MM NACL, 5MM MGCL2, 1MM RIBULOSE-1,5- BISPHOSPHATE, 1MM ATP/ATPGAMMAS |
| Specimen | Conc.: 0.07 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: YES / Vitrification applied: NO |
| EM staining | Type: NEGATIVE / Material: uranyl acetate |
| Specimen support | Details: CARBON |
- Electron microscopy imaging
Electron microscopy imaging
| Microscopy | Model: FEI TECNAI 12 / Date: Nov 19, 2010 |
|---|---|
| Electron gun | Electron source: LAB6 / Accelerating voltage: 120 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELDBright-field microscopy / Nominal magnification: 90600 X / Calibrated magnification: 90600 X / Nominal defocus max: 1800 nm / Nominal defocus min: 260 nm / Cs: 2 mm |
| Image recording | Electron dose: 20 e/Å2 / Film or detector model: FEI EAGLE (2k x 2k) |
| Image scans | Num. digital images: 12 |
| Radiation wavelength | Relative weight: 1 |
- Processing
Processing
| EM software |
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Details: PHASE FLIPPING, EACH PARTICLE | ||||||||||||
| Symmetry | Point symmetry: C6 (6 fold cyclic) | ||||||||||||
| 3D reconstruction | Method: ANGULAR RECONSTITUTION / Resolution: 21 Å / Num. of particles: 245 / Nominal pixel size: 3.308 Å / Actual pixel size: 3.308 Å Details: MODULE CONSISTING OF CHAIN A (204-296) AND CHAIN B (8-203) AS FOUND IN PDB 3SYL WAS MODELLED ONTO D2 P97 OF PDB 3CF3. SUBMISSION BASED ON EXPERIMENTAL DATA FROM EMDB EMD-1932. Symmetry type: POINT | ||||||||||||
| Atomic model building | Protocol: RIGID BODY FIT / Space: REAL / Details: METHOD--RIGID BODY REFINEMENT PROTOCOL--MANUAL | ||||||||||||
| Atomic model building | PDB-ID: 3SYL | ||||||||||||
| Refinement | Highest resolution: 21 Å | ||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 21 Å
|
