 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-1121: Mapping the structure and function of the E1 and E2 glycoproteins... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1121 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. | |||||||||
 Map data Map data | center of virus is where z=0 | |||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology informationicosahedral viral capsid, spike /  togavirin / T=4 icosahedral viral capsid / ubiquitin-like protein ligase binding / symbiont-mediated suppression of host toll-like receptor signaling pathway / clathrin-dependent endocytosis of virus by host cell / host cell cytoplasm / membrane fusion / membrane => GO:0016020 / symbiont entry into host cell ...icosahedral viral capsid, spike / togavirin / T=4 icosahedral viral capsid / ubiquitin-like protein ligase binding / symbiont-mediated suppression of host toll-like receptor signaling pathway / clathrin-dependent endocytosis of virus by host cell / host cell cytoplasm / membrane fusion / membrane => GO:0016020 / symbiont entry into host cell / serine-type endopeptidase activity / fusion of virus membrane with host endosome membrane / viral envelope / host cell nucleus / structural molecule activity / virion attachment to host cell / host cell plasma membrane / virion membrane / proteolysis / RNA binding / membrane / identical protein binding / plasma membrane / cytoplasm togavirin / T=4 icosahedral viral capsid / ubiquitin-like protein ligase binding / symbiont-mediated suppression of host toll-like receptor signaling pathway / clathrin-dependent endocytosis of virus by host cell / host cell cytoplasm / membrane fusion / membrane => GO:0016020 / symbiont entry into host cell ...icosahedral viral capsid, spike / togavirin / T=4 icosahedral viral capsid / ubiquitin-like protein ligase binding / symbiont-mediated suppression of host toll-like receptor signaling pathway / clathrin-dependent endocytosis of virus by host cell / host cell cytoplasm / membrane fusion / membrane => GO:0016020 / symbiont entry into host cell / serine-type endopeptidase activity / fusion of virus membrane with host endosome membrane / viral envelope / host cell nucleus / structural molecule activity / virion attachment to host cell / host cell plasma membrane / virion membrane / proteolysis / RNA binding / membrane / identical protein binding / plasma membrane / cytoplasmSimilarity search - Function | |||||||||
| Biological species |  Sindbis virus Sindbis virus | |||||||||
| Method | single particle reconstruction / cryo EM / negative staining / Resolution: 9.0 Å | |||||||||
 Authors Authors | Mukhopadhyay S / Zhang W / Gabler S / Chipman PR / Strauss EG / Strauss JH / Baker TS / Kuhn RJ / Rossmann MG | |||||||||
 Citation Citation | Journal: Structure / Year: 2006 Title: Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Authors: Suchetana Mukhopadhyay / Wei Zhang / Stefan Gabler / Paul R Chipman / Ellen G Strauss / James H Strauss / Timothy S Baker / Richard J Kuhn / Michael G Rossmann /  Abstract: The 9 A resolution cryo-electron microscopy map of Sindbis virus presented here provides structural information on the polypeptide topology of the E2 protein, on the interactions between the E1 and ...The 9 A resolution cryo-electron microscopy map of Sindbis virus presented here provides structural information on the polypeptide topology of the E2 protein, on the interactions between the E1 and E2 glycoproteins in the formation of a heterodimer, on the difference in conformation of the two types of trimeric spikes, on the interaction between the transmembrane helices of the E1 and E2 proteins, and on the conformational changes that occur when fusing with a host cell. The positions of various markers on the E2 protein established the approximate topology of the E2 structure. The largest conformational differences between the icosahedral surface spikes at icosahedral 3-fold and quasi-3-fold positions are associated with the monomers closest to the 5-fold axes. The long E2 monomers, containing the cell receptor recognition motif at their extremities, are shown to rotate by about 180 degrees and to move away from the center of the spikes during fusion. | |||||||||
| History |
|
- Structure visualization
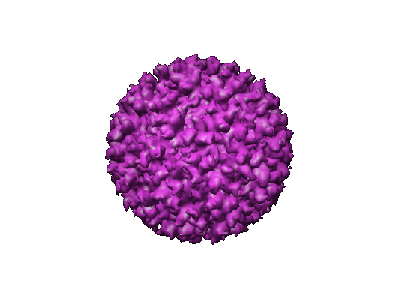
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1121.map.gz | 125.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1121-v30.xmlemd-1121.xml | 12.9 KB 12.9 KB | Display Display | EMDB header |
| Images |  1121.gif 1121.gif | 33.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1121ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1121 http://ftp.pdbj.org/pub/emdb/structures/EMD-1121ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1121 | HTTPS FTP |
-Related structure data
| Related structure data |  1z8yMC  2xfbM  3muwM M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_1121.map.gz / Format: CCP4 / Size: 319.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | center of virus is where z=0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.78499 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Sindbis TE12 E2-N318Q
| Entire | Name: Sindbis TE12 E2-N318Q |
|---|---|
| Components |
|
-Supramolecule #1000: Sindbis TE12 E2-N318Q
| Supramolecule | Name: Sindbis TE12 E2-N318Q / type: sample / ID: 1000 Details: Sample is a single deglycosylated virus. For mutagenesis and purification, please see Pletnev et al. (2001) Cell. Virus contains 3 proteins (E1, E2, capsid), a lipid bilyer, and a positive-strand RNA genome. Number unique components: 1 |
|---|
-Supramolecule #1: Sindbis virus
| Supramolecule | Name: Sindbis virus / type: virus / ID: 1 / Name.synonym: Sindbis strain TE12 / Details: deglycosylated virus / NCBI-ID: 11034 / Sci species name: Sindbis virus / Virus type: VIRION / Virus isolate: STRAIN / Virus enveloped: Yes / Virus empty: No / Syn species name: Sindbis strain TE12 |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) / synonym: INVERTEBRATES Homo sapiens (human) / synonym: INVERTEBRATES |
| Molecular weight | Experimental: 52 MDa |
| Virus shell | Shell ID: 1 / Name: glycoprotein / Diameter: 710 Å / T number (triangulation number): 4 |
| Virus shell | Shell ID: 2 / Name: lipid bilayer / Diameter: 480 Å / T number (triangulation number): 4 |
| Virus shell | Shell ID: 3 / Name: capsid / Diameter: 410 Å / T number (triangulation number): 4 |
-Experimental details
-Structure determination
| Method | negative staining, cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 6 mg/mL |
|---|---|
| Buffer | pH: 7.4 / Details: 20 mM Tris-Cl, 200 mM NaCl, 0.1 mM EDTA |
| Staining | Type: NEGATIVE / Details: no staining |
| Grid | Details: holey carbon 400 mesh copper grid |
| Vitrification | Cryogen name: ETHANE / Instrument: HOMEMADE PLUNGER Details: Vitrification instrument: Purdue manufactured, gravity driven device. Vitrification carried out in hood. Timed resolved state: Vitrified immediately after blotting. / Method: standard methods |
- Electron microscopy
Electron microscopy
| Microscope | FEI/PHILIPS CM200T |
|---|---|
| Electron beam | Acceleration voltage: 200 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 39220 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.0 mm / Nominal defocus max: 2.58 µm / Nominal defocus min: 1.1 µm / Nominal magnification: 38000 |
| Sample stage | Specimen holder: Side entry liquid nitrogen-cooled cryo specimen holder Specimen holder model: GATAN LIQUID NITROGEN |
| Temperature | Min: 87 K / Max: 100 K |
| Alignment procedure | Legacy - Astigmatism: objective lens astigmatism was corrected at 100,000 times magnification |
| Details | Low dose |
| Date | Jun 21, 2000 |
| Image recording | Category: FILM / Film or detector model: KODAK SO-163 FILM / Digitization - Scanner: ZEISS SCAI / Digitization - Sampling interval: 7 µm / Number real images: 27 / Average electron dose: 18 e/Å2 / Details: optical density range is 0.33-1.35 / Od range: 1 / Bits/pixel: 8 |
| Tilt angle min | 0 |
| Tilt angle max | 0 |
-Image processing
| CTF correction | Details: CTF correction of each particle. |
|---|---|
| Final two d classification | Number classes: 3 |
| Final angle assignment | Details: Euler angles (theta, phi, omega) are defined as three successive rotations in a right hand coordinate system. First, the viewer is rotated counterclockwise around the z-axis (angle 'phi') ...Details: Euler angles (theta, phi, omega) are defined as three successive rotations in a right hand coordinate system. First, the viewer is rotated counterclockwise around the z-axis (angle 'phi') and then rotated counterclockwise around the new y-axis (angle 'theta') and rotate clockwise around the new z-axis (angle 'omega'). |
| Final reconstruction | Applied symmetry - Point group: I (icosahedral) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 9.0 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: Purdue Suite Details: resolution determined by splitting data set into two groups. Number images used: 7085 |
| Details | The particles were selected interactively at the computer terminal. |
-Atomic model buiding 1
| Initial model | PDB ID: |
|---|---|
| Software | Name: EMFIT |
| Details | Protocol: Rigid body. each protein in the asymmetric unit was fitting individually. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: sumf |
| Output model | PDB-1z8y: PDB-2xfb: PDB-3muw: |

-Atomic model buiding 2
| Initial model | PDB ID: |
|---|---|
| Software | Name: EMFIT |
| Details | Protocol: Rigid body. each protein in the asymmetric unit was fitting individually. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: sumf |
| Output model | PDB-1z8y: PDB-2xfb: PDB-3muw: |

