Journal: J Biol Chem / Year: 2014 Title: Systematic comparison of molecular conformations of H+,K+-ATPase reveals an important contribution of the A-M2 linker for the luminal gating. Authors: Kazuhiro Abe / Kazutoshi Tani / Yoshinori Fujiyoshi / Abstract: Gastric H(+),K(+)-ATPase, an ATP-driven proton pump responsible for gastric acidification, is a molecular target for anti-ulcer drugs. Here we show its cryo-electron microscopy (EM) structure in an ...Gastric H(+),K(+)-ATPase, an ATP-driven proton pump responsible for gastric acidification, is a molecular target for anti-ulcer drugs. Here we show its cryo-electron microscopy (EM) structure in an E2P analog state, bound to magnesium fluoride (MgF), and its K(+)-competitive antagonist SCH28080, determined at 7 Å resolution by electron crystallography of two-dimensional crystals. Systematic comparison with other E2P-related cryo-EM structures revealed that the molecular conformation in the (SCH)E2·MgF state is remarkably distinguishable. Although the azimuthal position of the A domain of the (SCH)E2·MgF state is similar to that in the E2·AlF (aluminum fluoride) state, in which the transmembrane luminal gate is closed, the arrangement of transmembrane helices in the (SCH)E2·MgF state shows a luminal-open conformation imposed on by bound SCH28080 at its luminal cavity, based on observations of the structure in the SCH28080-bound E2·BeF (beryllium fluoride) state. The molecular conformation of the (SCH)E2·MgF state thus represents a mixed overall structure in which its cytoplasmic and luminal half appear to be independently modulated by a phosphate analog and an antagonist bound to the respective parts of the enzyme. Comparison of the molecular conformations revealed that the linker region connecting the A domain and the transmembrane helix 2 (A-M2 linker) mediates the regulation of luminal gating. The mechanistic rationale underlying luminal gating observed in H(+),K(+)-ATPase is consistent with that observed in sarcoplasmic reticulum Ca(2+)-ATPase and other P-type ATPases and is most likely conserved for the P-type ATPase family in general.
Resolution: 8→128.95 Å / Rfactor Rfree error: 0.043 / Data cutoff high absF: 876135.35 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: BULK SOLVENT MODEL USED SUBMISSION BASED ON EXPERIMENTAL DATA FROM EMDB EMD-2759. (DEPOSITION ID: 12786).
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords TRANSPORT PROTEIN / POTASSIUM-TRANSPORTING ATPASE
TRANSPORT PROTEIN / POTASSIUM-TRANSPORTING ATPASE Function and homology information
Function and homology information
 Authors
Authors Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj
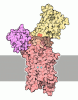

 Assembly
Assembly
 Sample preparation
Sample preparation Processing
Processing