 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3453: Cryo-EM structure of human p53 bound to a molecular support struc... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3453 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of human p53 bound to a molecular support structure made of DNA origami. | |||||||||
 Map data Map data | None | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 15.0 Å | |||||||||
 Authors Authors | Martin TG / Bharat TAM / Joerger AC / Bai X / Praetorius F / Fersht AR / Dietz H / Scheres SHW | |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2016 Title: Design of a molecular support for cryo-EM structure determination. Authors: Thomas G Martin / Tanmay A M Bharat / Andreas C Joerger / Xiao-Chen Bai / Florian Praetorius / Alan R Fersht / Hendrik Dietz / Sjors H W Scheres /   Abstract: Despite the recent rapid progress in cryo-electron microscopy (cryo-EM), there still exist ample opportunities for improvement in sample preparation. Macromolecular complexes may disassociate or ...Despite the recent rapid progress in cryo-electron microscopy (cryo-EM), there still exist ample opportunities for improvement in sample preparation. Macromolecular complexes may disassociate or adopt nonrandom orientations against the extended air-water interface that exists for a short time before the sample is frozen. We designed a hollow support structure using 3D DNA origami to protect complexes from the detrimental effects of cryo-EM sample preparation. For a first proof-of-principle, we concentrated on the transcription factor p53, which binds to specific DNA sequences on double-stranded DNA. The support structures spontaneously form monolayers of preoriented particles in a thin film of water, and offer advantages in particle picking and sorting. By controlling the position of the binding sequence on a single helix that spans the hollow support structure, we also sought to control the orientation of individual p53 complexes. Although the latter did not yet yield the desired results, the support structures did provide partial information about the relative orientations of individual p53 complexes. We used this information to calculate a tomographic 3D reconstruction, and refined this structure to a final resolution of ∼15 Å. This structure settles an ongoing debate about the symmetry of the p53 tetramer bound to DNA. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3453.map.gz | 1.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3453-v30.xmlemd-3453.xml | 12.5 KB 12.5 KB | Display Display | EMDB header |
| Images | 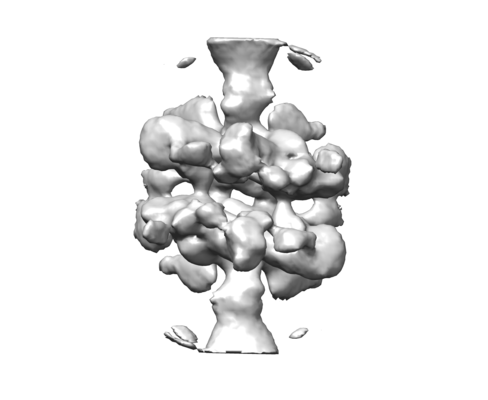 emd_3453.png emd_3453.png | 45.6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3453ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3453 http://ftp.pdbj.org/pub/emdb/structures/EMD-3453ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3453 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_3453.map.gz / Format: CCP4 / Size: 2.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | None | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.76 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Tetramer of truncated human p53 (residues 1-360) bound to dsDNA
| Entire | Name: Tetramer of truncated human p53 (residues 1-360) bound to dsDNA |
|---|---|
| Components |
|
-Supramolecule #1: Tetramer of truncated human p53 (residues 1-360) bound to dsDNA
| Supramolecule | Name: Tetramer of truncated human p53 (residues 1-360) bound to dsDNA type: complex / ID: 1 / Parent: 0 Details: The dsDNA to which p53 was bound is part of a large DNA origami support structure that was used in the structure determination process. |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism:  Escherichia coli (E. coli) / Recombinant plasmid: Modified pET24a expression vecto Escherichia coli (E. coli) / Recombinant plasmid: Modified pET24a expression vecto |
| Molecular weight | Theoretical: 160 KDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 |
|---|---|
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE |
| Details | P53 tetramers were bound to large DNA origami support structures. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated defocus max: 5.0 µm / Calibrated defocus min: 1.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy |
| Specialist optics | Energy filter - Name: GIF / Energy filter - Lower energy threshold: -20 eV / Energy filter - Upper energy threshold: +20 eV |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Frames/image: 1-20 / Number grids imaged: 7 / Number real images: 2562 / Average exposure time: 16.0 sec. / Average electron dose: 38.0 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 272914 |
|---|---|
| CTF correction | Software - Name: Gctf |
| Startup model | Type of model: OTHER Details: Tomographic reconstruction from angles experimentally determined by DNA origami support alignment. |
| Initial angle assignment | Type: OTHER / Software - Name: RELION (ver. 1.4) Details: Tomographic angles experimentally determined by DNA origami support alignment. |
| Final angle assignment | Type: PROJECTION MATCHING / Software - Name: RELION (ver. 1.4) |
| Final reconstruction | Applied symmetry - Point group: C2 (2 fold cyclic) / Resolution.type: BY AUTHOR / Resolution: 15.0 Å / Resolution method: OTHER / Software - Name: RELION (ver. 2) / Software - details: Beta Details: FSC=0.143 indicates a resolution around 10A.However, visual inspection of the map indicates this may be a bit too high. As some developmental image processing procedures were used to take ...Details: FSC=0.143 indicates a resolution around 10A.However, visual inspection of the map indicates this may be a bit too high. As some developmental image processing procedures were used to take into account the experimental information provided by the DNA origami support structures, we chose to stick to a more conservative 15A resolution estimate. Number images used: 9271 |
