 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qz5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of rabbit actin in complex with kabiramide C | ||||||
 Components Components | Actin, alpha skeletal muscle | ||||||
 Keywords Keywords | STRUCTURAL PROTEIN / actin / trisoxazole / toxin / kabiramide C | ||||||
| Function / homology |  Function and homology information Function and homology informationcytoskeletal motor activator activity / tropomyosin binding / myosin heavy chain binding / mesenchyme migration / troponin I binding / actin filament bundle / filamentous actin / actin filament bundle assembly / skeletal muscle thin filament assembly / striated muscle thin filament ...cytoskeletal motor activator activity / tropomyosin binding / myosin heavy chain binding / mesenchyme migration / troponin I binding / actin filament bundle / filamentous actin / actin filament bundle assembly / skeletal muscle thin filament assembly / striated muscle thin filament / skeletal muscle myofibril / actin monomer binding / skeletal muscle fiber development / stress fiber / titin binding / actin filament polymerization / filopodium / actin filament / Hydrolases; Acting on acid anhydrides; Acting on acid anhydrides to facilitate cellular and subcellular movement / calcium-dependent protein binding / lamellipodium / cell body / hydrolase activity / protein domain specific binding / calcium ion binding / positive regulation of gene expression / magnesium ion binding / ATP binding / identical protein binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Oryctolagus cuniculus (rabbit) Oryctolagus cuniculus (rabbit) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | ||||||
 Authors Authors | Klenchin, V.A. / Allingham, J.S. / King, R. / Tanaka, J. / Marriott, G. / Rayment, I. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 2003 Title: Trisoxazole macrolide toxins mimic the binding of actin-capping proteins to actin Authors: Klenchin, V.A. / Allingham, J.S. / King, R. / Tanaka, J. / Marriott, G. / Rayment, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qz5.cif.gz | 90.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qz5.ent.gz | 74.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1qz5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qz/1qz5ftp://data.pdbj.org/pub/pdb/validation_reports/qz/1qz5 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / Alpha-actin 1 Mass: 41875.633 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Oryctolagus cuniculus (rabbit) / References: UniProt: P68135 |
|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #3: Chemical | ChemComp-ATP / Adenosine triphosphate  Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM |
| #4: Chemical | ChemComp-KAB / 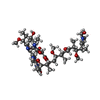  Mass: 946.134 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C48H75N5O14 Mass: 946.134 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C48H75N5O14 |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 357 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 357 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.21 Å3/Da / Density % sol: 44.43 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: batch / pH: 5.5 Details: Crystals of the actin-kabiramide C complex were grown by small-scale batch by mixing equal volumes of the complex and 100 mM MES, 18% (w/v) polyethylene glycol 1500, 12% (w/v) 1,6- ...Details: Crystals of the actin-kabiramide C complex were grown by small-scale batch by mixing equal volumes of the complex and 100 mM MES, 18% (w/v) polyethylene glycol 1500, 12% (w/v) 1,6-hexanediol, 100 mM CaCl2, 1 mM sodium azide, 1 mM TCEP, pH 5.5, Batch, temperature 293K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: batch method | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 0.9 Å / Beamline: 14-BM-C / Wavelength: 0.9 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Aug 23, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→54 Å / Num. all: 65708 / Num. obs: 65708 / % possible obs: 98.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 8.3 % / Biso Wilson estimate: 11 Å2 / Rmerge(I) obs: 0.044 / Net I/σ(I): 47.1 |
| Reflection shell | Resolution: 1.45→1.5 Å / Rmerge(I) obs: 0.123 / Mean I/σ(I) obs: 13.9 / Num. unique all: 6310 / % possible all: 10 |
| Reflection | *PLUS Lowest resolution: 30 Å / Num. measured all: 542898 |
| Reflection shell | *PLUS % possible obs: 95.7 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.45→30 Å / Cor.coef. Fo:Fc: 0.964 / Cor.coef. Fo:Fc free: 0.959 / SU B: 0.984 / SU ML: 0.039 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.067 / ESU R Free: 0.065 / Stereochemistry target values: Engh & Huber / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.301 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.45→1.5 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 30 Å / % reflection Rfree: 5 % / Rfactor Rfree: 0.188 / Rfactor Rwork: 0.169 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rwork: 0.187 |
