 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8220 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
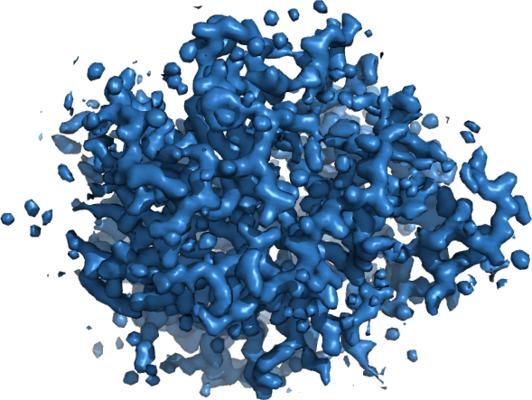
| Title | MicroED structure of trypsin at 1.7 A resolution | |||||||||
 Map data Map data | Trypsin | |||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / extracellular space / metal ion binding Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / extracellular space / metal ion bindingSimilarity search - Function | |||||||||
| Biological species |  Bos taurus (cattle) / Bovine (cattle) Bos taurus (cattle) / Bovine (cattle) | |||||||||
| Method | electron crystallography / cryo EM / Resolution: 1.7 Å | |||||||||
 Authors Authors | de la Cruz MJ / Hattne J / Shi D / Seidler P / Rodriguez J / Reyes FE / Sawaya MR / Cascio D / Eisenberg D / Gonen T | |||||||||
 Citation Citation | Journal: Nat Methods / Year: 2017 Title: Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED. Authors: M Jason de la Cruz / Johan Hattne / Dan Shi / Paul Seidler / Jose Rodriguez / Francis E Reyes / Michael R Sawaya / Duilio Cascio / Simon C Weiss / Sun Kyung Kim / Cynthia S Hinck / Andrew P ...Authors: M Jason de la Cruz / Johan Hattne / Dan Shi / Paul Seidler / Jose Rodriguez / Francis E Reyes / Michael R Sawaya / Duilio Cascio / Simon C Weiss / Sun Kyung Kim / Cynthia S Hinck / Andrew P Hinck / Guillermo Calero / David Eisenberg / Tamir Gonen /  Abstract: Traditionally, crystallographic analysis of macromolecules has depended on large, well-ordered crystals, which often require significant effort to obtain. Even sizable crystals sometimes suffer from ...Traditionally, crystallographic analysis of macromolecules has depended on large, well-ordered crystals, which often require significant effort to obtain. Even sizable crystals sometimes suffer from pathologies that render them inappropriate for high-resolution structure determination. Here we show that fragmentation of large, imperfect crystals into microcrystals or nanocrystals can provide a simple path for high-resolution structure determination by the cryoEM method MicroED and potentially by serial femtosecond crystallography. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8220.map.gz | 3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8220-v30.xmlemd-8220.xml | 15.5 KB 15.5 KB | Display Display | EMDB header |
| Images | 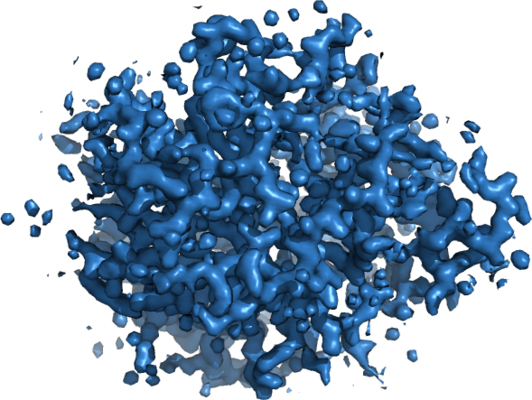 emd_8220.png emd_8220.png | 269 KB | ||
| Filedesc structureFactors | emd_8220_sf.cif.gz | 1.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8220ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8220 http://ftp.pdbj.org/pub/emdb/structures/EMD-8220ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8220 | HTTPS FTP |
-Related structure data
| Related structure data |  5k7rMC  8216C  8217C  8218C  8219C  8221C  8222C  8472C  5k7nC  5k7oC  5k7pC  5k7qC  5k7sC  5k7tC  5ty4C C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_8220.map.gz / Format: CCP4 / Size: 3.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Trypsin | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X: 0.554 Å / Y: 0.56429 Å / Z: 0.57743 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Trypsin
| Entire | Name: Trypsin |
|---|---|
| Components |
|
-Supramolecule #1: Trypsin
| Supramolecule | Name: Trypsin / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Bos taurus (cattle) |
| Molecular weight | Theoretical: 23.354 KDa |
-Macromolecule #1: Cationic trypsin
| Macromolecule | Name: Cationic trypsin / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO / EC number: trypsin |
|---|---|
| Source (natural) | Organism: Bovine (cattle) |
| Molecular weight | Theoretical: 23.324287 KDa |
| Sequence | String: IVGGYTCGAN TVPYQVSLNS GYHFCGGSLI NSQWVVSAAH CYKSGIQVRL GEDNINVVEG NEQFISASKS IVHPSYNSNT LNNDIMLIK LKSAASLNSR VASISLPTSC ASAGTQCLIS GWGNTKSSGT SYPDVLKCLK APILSDSSCK SAYPGQITSN M FCAGYLEG ...String: IVGGYTCGAN TVPYQVSLNS GYHFCGGSLI NSQWVVSAAH CYKSGIQVRL GEDNINVVEG NEQFISASKS IVHPSYNSNT LNNDIMLIK LKSAASLNSR VASISLPTSC ASAGTQCLIS GWGNTKSSGT SYPDVLKCLK APILSDSSCK SAYPGQITSN M FCAGYLEG GKDSCQGDSG GPVVCSGKLQ GIVSWGSGCA QKNKPGVYTK VCNYVSWIKQ TIASN |
-Macromolecule #2: CALCIUM ION
| Macromolecule | Name: CALCIUM ION / type: ligand / ID: 2 / Number of copies: 2 / Formula: CA |
|---|---|
| Molecular weight | Theoretical: 40.078 Da |
-Macromolecule #3: water
| Macromolecule | Name: water / type: ligand / ID: 3 / Number of copies: 195 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | electron crystallography |
| Aggregation state | 3D array |
-Sample preparation
| Buffer | pH: 6.5 Component:
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Electron beam | Acceleration voltage: 200 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: DIFFRACTION / Camera length: 1500 mm |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Image recording | Film or detector model: TVIPS TEMCAM-F416 (4k x 4k) / Digitization - Dimensions - Width: 2048 pixel / Digitization - Dimensions - Height: 2048 pixel / Digitization - Sampling interval: 0.0311999992 µm / Number grids imaged: 3 / Number real images: 1527 / Number diffraction images: 1527 / Average exposure time: 4.1 sec. / Average electron dose: 0.004 e/Å2 |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
-Image processing
| Crystal parameters | Unit cell - A: 53.12 Å / Unit cell - B: 56.08 Å / Unit cell - C: 64.38 Å / Unit cell - γ: 90 ° / Unit cell - α: 90 ° / Unit cell - β: 90 ° / Space group: P 21 21 21 |
|---|---|
| Crystallography statistics | Number intensities measured: 145833 / Number structure factors: 23542 / Fourier space coverage: 73.8 / R sym: 0.773 / R merge: 0.773 / Overall phase error: 28.86 / Overall phase residual: 40.4 / Phase error rejection criteria: 0 / High resolution: 1.5 Å / Shell - Shell ID: 1 / Shell - High resolution: 1.7 Å / Shell - Low resolution: 1.79 Å / Shell - Number structure factors: 1737 / Shell - Phase residual: 60.7 / Shell - Fourier space coverage: 56.2 / Shell - Multiplicity: 3.1 |
| Molecular replacement | Software - Name: MOLREP (ver. 11.4.05) / Software - details: Starting model PDB ID 2ptn |
| Symmetry determination software list | Software - Name: POINTLESS (ver. 1.10.21) |
| Final reconstruction | Resolution.type: BY AUTHOR / Resolution: 1.7 Å / Resolution method: DIFFRACTION PATTERN/LAYERLINES |
| Merging software list | Software - Name: AIMLESS (ver. 0.5.25) |

