 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-5529 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
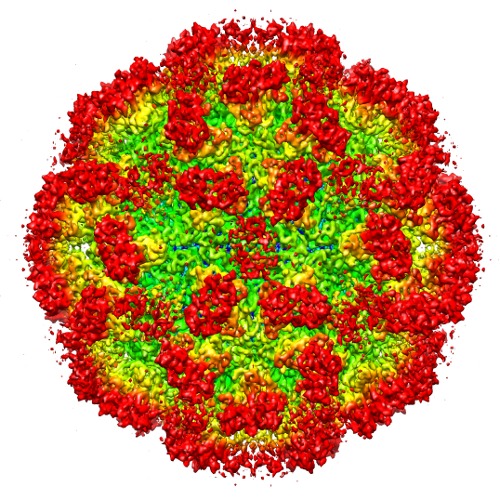
| Title | 6.3 A Cryo-EM Structure of a Novel Calicivirus, Tulane Virus | |||||||||
 Map data Map data | Reconstruction of TV virion | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Tulane virus / calicivirus / conformational flexibility / single particle cryo-EM / 3-D reconstruction | |||||||||
| Biological species |  Tulane virus Tulane virus | |||||||||
| Method | single particle reconstruction / cryo EM / negative staining / Resolution: 6.3 Å | |||||||||
 Authors Authors | Yu G / Zhang D / Guo F / Tan M / Jiang X / Jiang W | |||||||||
 Citation Citation | Journal: PLoS One / Year: 2013 Title: Cryo-EM structure of a novel calicivirus, Tulane virus. Authors: Guimei Yu / Dongsheng Zhang / Fei Guo / Ming Tan / Xi Jiang / Wen Jiang /  Abstract: Tulane virus (TV) is a newly isolated cultivatable calicivirus that infects juvenile rhesus macaques. Here we report a 6.3 Å resolution cryo-electron microscopy structure of the TV virion. The TV ...Tulane virus (TV) is a newly isolated cultivatable calicivirus that infects juvenile rhesus macaques. Here we report a 6.3 Å resolution cryo-electron microscopy structure of the TV virion. The TV virion is about 400 Å in diameter and consists of a T = 3 icosahedral protein capsid enclosing the RNA genome. 180 copies of the major capsid protein VP1 (∼57 KDa) are organized into two types of dimers A/B and C/C and form a thin, smooth shell studded with 90 dimeric protrusions. The overall capsid organization and the capsid protein fold of TV closely resemble that of other caliciviruses, especially of human Norwalk virus, the prototype human norovirus. These close structural similarities support TV as an attractive surrogate for the non-cultivatable human noroviruses. The most distinctive feature of TV is that its C/C dimers are in a highly flexible conformation with significantly reduced interactions between the shell (S) domain and the protruding (P) domain of VP1. A comparative structural analysis indicated that the P domains of TV C/C dimers were much more flexible than those of other caliciviruses. These observations, combined with previous studies on other caliciviruses, led us to hypothesize that the enhanced flexibility of C/C dimer P domains are likely required for efficient calicivirus-host cell interactions and the consequent uncoating and genome release. Residues in the S-P1 hinge between the S and P domain may play a critical role in the flexibility of P domains of C/C dimers. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_5529.map.gz | 55.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-5529-v30.xmlemd-5529.xml | 12.4 KB 12.4 KB | Display Display | EMDB header |
| Images | emd_5529.tif | 732.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-5529ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5529 http://ftp.pdbj.org/pub/emdb/structures/EMD-5529ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5529 | HTTPS FTP |
-Validation report
| Summary document | emd_5529_validation.pdf.gz | 79.3 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_5529_full_validation.pdf.gz | 78.4 KB | Display | |
| Data in XML | emd_5529_validation.xml.gz | 493 B | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-5529ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-5529 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_5529.map.gz / Format: CCP4 / Size: 162.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of TV virion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.74 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Tulane virus
| Entire | Name: Tulane virus |
|---|---|
| Components |
|
-Supramolecule #1000: Tulane virus
| Supramolecule | Name: Tulane virus / type: sample / ID: 1000 Oligomeric state: One Tulane virus has 90 dimers forming its icosahedral capsid (T=3). Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 10.4 MDa |
-Supramolecule #1: Tulane virus
| Supramolecule | Name: Tulane virus / type: virus / ID: 1 / NCBI-ID: 512169 / Sci species name: Tulane virus / Database: NCBI / Virus type: VIRION / Virus isolate: SPECIES / Virus enveloped: No / Virus empty: No |
|---|---|
| Host (natural) | Organism:  |
| Molecular weight | Theoretical: 10.4 MDa |
| Virus shell | Shell ID: 1 / Name: VP1 / Diameter: 400 Å / T number (triangulation number): 3 |
-Experimental details
-Structure determination
| Method | negative staining, cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.4 Details: 137mM NaCl, 2.7mM KCl, 10mM Na2HPO4, 2mM KH2PO4, pH 7.4 |
|---|---|
| Staining | Type: NEGATIVE Details: Grids with sample floated on 2% uranyl acetate for 30 seconds. |
| Grid | Details: 400 mesh copper grid with one lacy carbon layer and one layer of ultra thin carbon on top. |
| Vitrification | Cryogen name: ETHANE / Chamber temperature: 85 K / Instrument: HOMEMADE PLUNGER |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 80 K / Max: 85 K / Average: 80 K |
| Alignment procedure | Legacy - Astigmatism: Objective lens astigmatism was corrected at 250,000 magnification using quadrupole stigmator. Legacy - Electron beam tilt params: 0 |
| Date | Jul 29, 2011 |
| Image recording | Category: FILM / Film or detector model: KODAK SO-163 FILM / Digitization - Scanner: NIKON SUPER COOLSCAN 9000 / Digitization - Sampling interval: 6.35 µm / Number real images: 190 / Average electron dose: 25 e/Å2 / Bits/pixel: 16 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 36475 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 4.891 µm / Nominal defocus min: 1.347 µm / Nominal magnification: 37000 |
| Sample stage | Specimen holder: Liquid nitrogen cooled / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Details | 4702 Tulane virus particles were selected using combined automated selection with ethan program and manual screening with boxer program in EMAN. The microscope contrast transfer function parameters for each micrograph were first determined using an automated fitting method and then manually verified/corrected using EMAN ctfit graphic program. The entire TV dataset was divided into two halves and processed independently for all the subsequent steps including construction of initial model, 2-D alignment and 3-D reconstruction. De novo initial models were constructed using the random model method in which random particle orientations were assigned and subsequently refined iteratively until convergence. The iterative refinement process including particle alignment and 3-D icosahedral reconstruction was performed using an in-house developed program jspr.py utilizing the EMAN/EMAN2 programs and library functions. The resolution was determined based on the 0.143 cutoff criterion for two truly independent reconstructions. |
|---|---|
| CTF correction | Details: each particle |
| Final reconstruction | Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 6.3 Å / Resolution method: OTHER / Software - Name: jspr.py, EMAN, EMAN2 / Number images used: 4338 |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C |
|---|---|
| Software | Name: Chimera |
| Details | Protocol: rigid body. The three chains from 1IHM were first fitted into TV density as a whole rigid body and then divided into dimers, specific chains, domains, and subdomains and fitted. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: Correlation |

