 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-12815 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Hepatitis B core protein -low secretion phenotype P5T | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology informationvirus-mediated perturbation of host defense response => GO:0019049 / microtubule-dependent intracellular transport of viral material towards nucleus / T=4 icosahedral viral capsid / : / viral penetration into host nucleus / host cell cytoplasm / symbiont entry into host cell / structural molecule activity /  DNA binding / RNA binding / extracellular region DNA binding / RNA binding / extracellular regionSimilarity search - Function | |||||||||
| Biological species |   Hepatitis B virus Hepatitis B virus | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.2 Å | |||||||||
 Authors Authors | Bottcher B / Makbul C | |||||||||
| Funding support |  Germany, 1 items Germany, 1 items
| |||||||||
 Citation Citation | Journal: Microorganisms / Year: 2021 Title: Conformational Plasticity of Hepatitis B Core Protein Spikes Promotes Peptide Binding Independent of the Secretion Phenotype. Authors: Cihan Makbul / Vladimir Khayenko / Hans Michael Maric / Bettina Böttcher / Abstract: Hepatitis B virus is a major human pathogen, which forms enveloped virus particles. During viral maturation, membrane-bound hepatitis B surface proteins package hepatitis B core protein capsids. This ...Hepatitis B virus is a major human pathogen, which forms enveloped virus particles. During viral maturation, membrane-bound hepatitis B surface proteins package hepatitis B core protein capsids. This process is intercepted by certain peptides with an "LLGRMKG" motif that binds to the capsids at the tips of dimeric spikes. With microcalorimetry, electron cryo microscopy and peptide microarray-based screens, we have characterized the structural and thermodynamic properties of peptide binding to hepatitis B core protein capsids with different secretion phenotypes. The peptide "GSLLGRMKGA" binds weakly to hepatitis B core protein capsids and mutant capsids with a premature (F97L) or low-secretion phenotype (L60V and P5T). With electron cryo microscopy, we provide novel structures for L60V and P5T and demonstrate that binding occurs at the tips of the spikes at the dimer interface, splaying the helices apart independent of the secretion phenotype. Peptide array screening identifies "SLLGRM" as the core binding motif. This shortened motif binds only to one of the two spikes in the asymmetric unit of the capsid and induces a much smaller conformational change. Altogether, these comprehensive studies suggest that the tips of the spikes act as an autonomous binding platform that is unaffected by mutations that affect secretion phenotypes. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_12815.map.gz | 228.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-12815-v30.xmlemd-12815.xml | 18.1 KB 18.1 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_12815_fsc.xml | 15.6 KB | Display | FSC data file |
| Images |  emd_12815.png emd_12815.png | 198.7 KB | ||
| Others | emd_12815_half_map_1.map.gzemd_12815_half_map_2.map.gz | 260 MB 260.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-12815ftp://ftp.pdbj.org/pub/emdb/structures/EMD-12815 http://ftp.pdbj.org/pub/emdb/structures/EMD-12815ftp://ftp.pdbj.org/pub/emdb/structures/EMD-12815 | HTTPS FTP |
-Related structure data
| Related structure data |  7ocwMC  7ocoC  7od4C  7od6C  7od7C  7od8C  7oenC  7oevC  7oewC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |

















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_12815.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Voxel size | X=Y=Z: 1.0635 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
-Half map: #1
| File | emd_12815_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
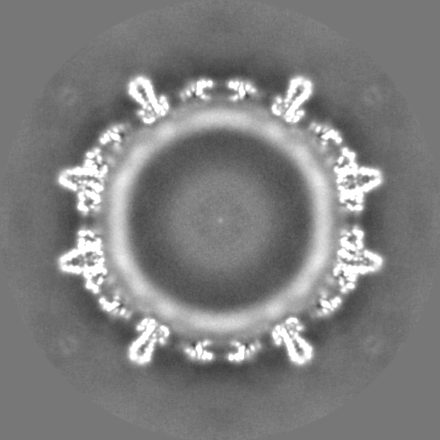
| Projections & Slices |
| ||||||||||||
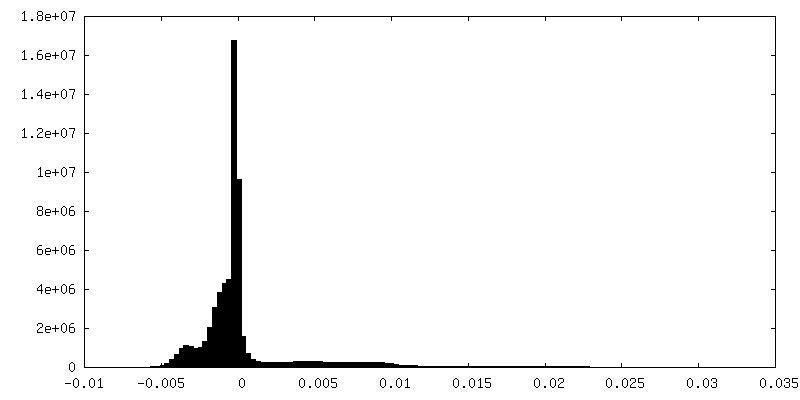
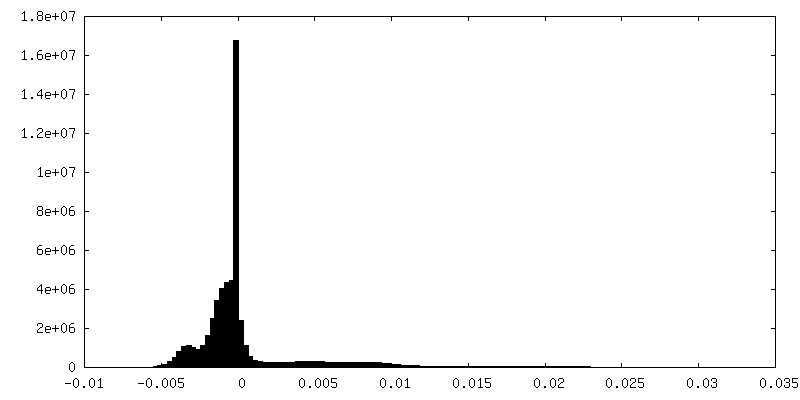
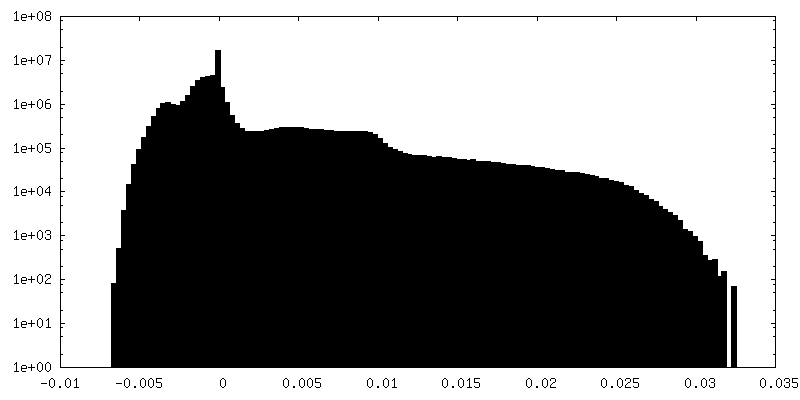
| Density Histograms |
 Z
Z Y
Y X
X






-Half map: #2
| File | emd_12815_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : Hepatitis B virus
| Entire | Name: Hepatitis B virus |
|---|---|
| Components |
|
-Supramolecule #1: Hepatitis B virus
| Supramolecule | Name: Hepatitis B virus / type: virus / ID: 1 / Parent: 0 / Macromolecule list: all / NCBI-ID: 10407 / Sci species name: Hepatitis B virus / Sci species strain: genotype D subtype ayw / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: STRAIN / Virus enveloped: No / Virus empty: No |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Host system | Organism:  Escherichia coli (E. coli) Escherichia coli (E. coli) |
| Molecular weight | Theoretical: 4.8 MDa |
| Virus shell | Shell ID: 1 / Name: capsid / Diameter: 360.0 Å / T number (triangulation number): 4 |
-Macromolecule #1: Capsid protein
| Macromolecule | Name: Capsid protein / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Hepatitis B virus |
| Molecular weight | Theoretical: 21.150205 KDa |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: MDIDTYKEFG ATVELLSFLP SDFFPSVRDL LDTASALYRE ALESPEHCSP HHTALRQAIL CWGELMTLAT WVGVNLEDPA SRDLVVSYV NTNMGLKFRQ LLWFHISCLT FGRETVIEYL VSFGVWIRTP PAYRPPNAPI LSTLPETTVV RRRGRSPRRR T PSPRRRRS QSPRRRRSQS RESQC |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 |
|---|---|
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 0.029 kPa |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | TFS KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 1.4000000000000001 µm / Nominal defocus min: 0.8 µm / Nominal magnification: 75000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: INTEGRATING / Number grids imaged: 1 / Number real images: 2421 / Average exposure time: 2.5 sec. / Average electron dose: 40.0 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 169111 |
|---|---|
| CTF correction | Software - Name: CTFFIND (ver. 4) |
| Startup model | Type of model: EMDB MAP EMDB ID: |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) |
| Final reconstruction | Applied symmetry - Point group: I (icosahedral) / Resolution.type: BY AUTHOR / Resolution: 3.2 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 3.1) / Number images used: 61896 |
| FSC plot (resolution estimation) |  |

